| Brian Pratt responded: |
2023-01-05 16:32 |
That should be possible, yes. If the data is viewable in ProteoWizard's SeeMS viewer then Skyline should work with it just fine. What are you doing for a spectral library?
Thanks for using the Skyline support board!
Brian Pratt
|
| |
| higgi022 responded: |
2023-01-09 15:17 |
I have confirmed that I am able to load in the data and partially visualize a chromatogram.
I have tried two different ways to utilize a spectral library:
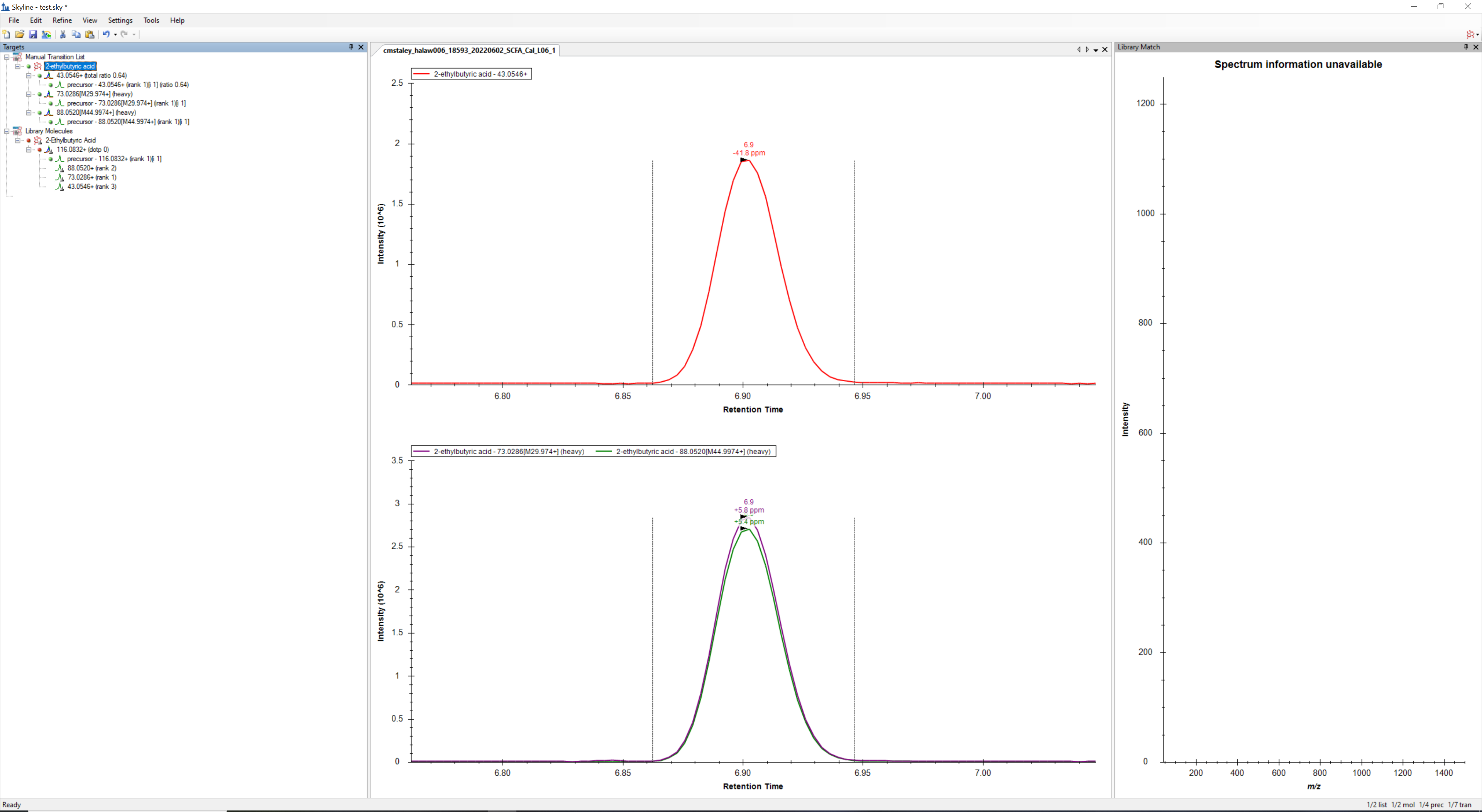
*1. Using an appropriately formatted MSP file. I am able to load in the file and add a compound to the experiment; however, Skyline constructs the compound using an MS/MS acquisition paradigm - a precursor and fragment ions. It attempts to draw a precursor trace, which doesn't exist in the GC-EI-MS results for that mass, and produces empty product ion traces, as there are no MS/MS in the experiment.
*2. Manually inserting a transition list. Through this route I specify multiple precursor m/z corresponding to the GC-EI-MS fragment ions, and duplicate the parameters for the product ion fields. Skyline draws all the desired traces as light and heavy version of the desired compounds. Unfortunately, I am not aware of any mechanism to specify the desired ratio of the "light" and "heavy" versions of the molecule to simulate spectral library filtering.
I attached a screen shot of my Skyline for reference. I think the second approach is very close to what I am looking for but I need to be able to use Dot Product ratios to filter my ions.
|
|
| |
| Brian Pratt responded: |
2023-01-09 16:33 |
I'll check with some GCMS Skyline users and try to come up with a better answer.
In the meantime, if you can provide your Skyline document and a sample data file, that may help us get where we want to be (knowing what settings are in use will be helpful). You can upload to http://skyline.ms/files.url or we can arrange something more private if you desire.
|
| |
| higgi022 responded: |
2023-01-10 08:22 |
I uploaded a zipped file containing the Skyline files and Agilent *.D file to the link you provided. The data represents testing material to assess system suitability, so no worries on privacy - use the data as you see fit. Additionally, I provided the MSP spectral library file and CSV transition list I used to create the molecule lists.
The provided raw files is a mix of 7 short chain fatty acids (SCFA) internal standards. The MSP file contains the expected intensities for 2-ethylbutyric acid fragment ions - this compound is not in the mix, but is normally the internal standard for this assay. There are a few siloxane contaminants coming from the inlet also observed in the file as well.
|
| |
| higgi022 responded: |
2023-02-04 08:57 |
@Brian Were you able to discover anything in regards to analyzing GC-QTOF results?
I've tried converting the Agilent *.D files to mzML/mzXML files and manually setting the spectrum level from MS1 to MS2. Unfortunately, Skyline does not recognize any chromatograms following this modification.
|
| |
| Nick Shulman responded: |
2023-02-04 09:16 |
If you send us your modified .mzML files we could tell you why Skyline is not giving you any chromatograms.
If you are going to be hand-editing mzML or mzXML files with a text editor, it is a good idea to create those files with the "--noindex" msconvert.exe commandline parameter (or, if you are using the MSConvert graphical user interface, uncheck the "Write index" checkbox). The reason is that if you make any changes to the number of bytes which are in the file, you would need to update the numbers in the index at the end of the XML file if it is there, so it is easier if that index does not exist.
Files which are less than 50MB can be attached to this support request. You can upload larger files here:
https://skyline.ms/files.url
It might also be helpful to see your new Skyline document. In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
-- Nick |
| |
| higgi022 responded: |
2023-02-04 09:38 |
Thank you for the suggestion Nick. Unfortunately, even after after removing the write index option I am still unable to read the modified mzXML file into Skyline.
Attached are the Skyline file, the original mzXML, and modified mzXML.
In the Skyline file are a compound I am hoping two find utilizing two different formats. The first attempts to use a spectral library approach. However, because there are no MS2 in the original file, I cannot monitor any of MS1 fragment ions. The second draws a chromatogram for the desired fragments but as heavy-labeled precursors. I am unaware any way to compare the precursor areas to a spectral library for dot-product filtering.
Please let me know if you have any questions. |
|
| |
| Nick Shulman responded: |
2023-02-04 10:24 |
There are a couple of things going wrong:
1. The "Acquisition Method" at "Settings > Transition Settings > Instrument" was set to "PRM". That means that Skyline will only match an MS2 spectrum to a precursor if the isolation m/z in the spectrum is very close to the precursor m/z. For "All Ions" (i.e. MSe), you should set the Acquisition Method to "DIA" and set the Isolation Scheme to "All Ions".
2. In order for Skyline to actually treat a MS2 spectrum in a .mzXML file as really MS2, it needs to have a <precursorMz> element. I have edited your mzXML file and inserted "<precursorMz>1000</precursorMz>" in front of every occurance of "<peaks " and then Skyline was happy.
3. Also, since all of the spectra in the file are now MS2, you should tell Skyline not to extract MS1. That means that you should set the "Isotope Peaks Included" at "Settings > Transition Settings > Full Scan" to "None". That will tell Skyline that precursor chromatograms should be extracted from the MS2 spectra instead of MS1.
I am attaching my new .mzXML and .sky.zip files where I made these changes and was able to get chromatograms.
-- Nick |
|
| |
| higgi022 responded: |
2023-02-04 10:32 |
Thank you for looking into this. Yes, I see how you modified the file and Skyline document to pull out the spectra for matching.
I will look into making a script to format my results to make them Skyline compatible.
Do you think it is worth providing native support for GC-QTOF results in Skyline? While I would certainly appreciate the functionality, I cannot say if there is a large enough demand to warrant an update at this time.
Thanks again Nick! |
| |
| Brian Pratt responded: |
2023-02-06 09:00 |
Let me look at this again, with the original data. We certainly don't want anyone to have to go through this kind of preprocessing, and indeed Skyline has been in is use with at least some varieties of GC-MS for quite some time now without any such issues.
Best regards,
Brian |
| |
| Brian Pratt responded: |
2023-02-06 12:38 |
One part of the problem seems to be our handling of the .MSP file. Was the example provided manipulated in any way, or is it straight out of the NIST library?
Thanks,
Brian |
| |
| Brian Pratt responded: |
2023-02-08 15:33 |
OK, I have identified and fixed a couple of issues that were preventing this from working smoothly. The next Skyline-Daily will handle this without any need for manipulation of the mass spec data - the data will be understood as GC-EI fragments even though it's tagged as MS1.
Thanks for alerting us to the problem.
Best regards,
Brian |
| |
 skyline_gc-ei-ms_attempt1.PNG
skyline_gc-ei-ms_attempt1.PNG