You should go to:
Settings > Transition Settings > Instrument
and change "Method match tolerance m/z" to a higher number.
Currently, you have that set to "0.2" which is not enough to accommodate the difference between the m/z's of the precursors in the document and what was actually isolated by the mass spectrometer.
I would recommend changing that number to 0.55, since that seems to be the largest difference between Skyline's precursor m/z and the spectra in your wiff file.
After you change the method match tolerance you should tell Skyline to extract chromatograms again by going to:
Edit > Manage Results > Reimport
-- Nick |
Thanks Nick. That seemed to help!
Now, I am wondering why the peaks do not look right (picture attached). Do you happen to know?
I also thought if I toggled through each Product Ion (on the Targets window), it would show a chromatogram for each Product Ion but it only seems to change chromatograms when clicking on each Precursor Ion. |
When you have multiple peptides selected like that, Skyline will show you chromatograms for all of the peptides at once. You aren't really expected to be get much information out of the multi-peptide view. You should select a particular peptide or precursor in order to see chromatograms that will help you figure out what is going wrong.
It sounds like you are saying that when you switch which transition is selected in the Targets tree, the displayed chromatogram is not changing. That probably means that you have "Transitions > Total" selected. You should right-click on the chromatogram and choose "Transitions > All" so that Skyline will display the separate transitions as different colored lines. (You can also choose "Transitions > Single" so that Skyline shows you just the one chromatogram for the currently selected transition).
I would also recommend that you choose "Transform > None". By default, Skyline shows interpolated chromatograms, which Skyline uses for peak picking and peak area calculations. Your chromatograms are very spiky, which tends to get smoothed out by the interpolation process, so you should make sure you are looking at the untransformed chromatogram.
When I look at the chromatograms for the individual precursors, I see that many of the chromatograms are very spiky, alternating between zero and other numbers. Often when that happens it is a good idea to click on points along the chromatogram so that Skyline brings up the full scan spectrum viewer. The full scan spectrum viewer will show you highlighted rectangles around your transition m/z's which indicate the m/z channel that Skyline summed across when extracting the chromatogram. Sometimes the chromatogram is spiky because the actual signal is just outside of the rectangle, and you can fix that by changing the mass accuracy to a higher number at "Settings > Transition Settings > Full Scan". In the case of your document, that does not seem to be happening, so I don't think that changing the mass accuracy will help things.
I think the reason that your chromatograms are spiky is that you have very weak signal and in different spectra it hovers around the value that is treated by the mass spectrometer as zero.
-- Nick |
 Screenshot of Data Import_Chromatogram information unavailable.JPG
Screenshot of Data Import_Chromatogram information unavailable.JPG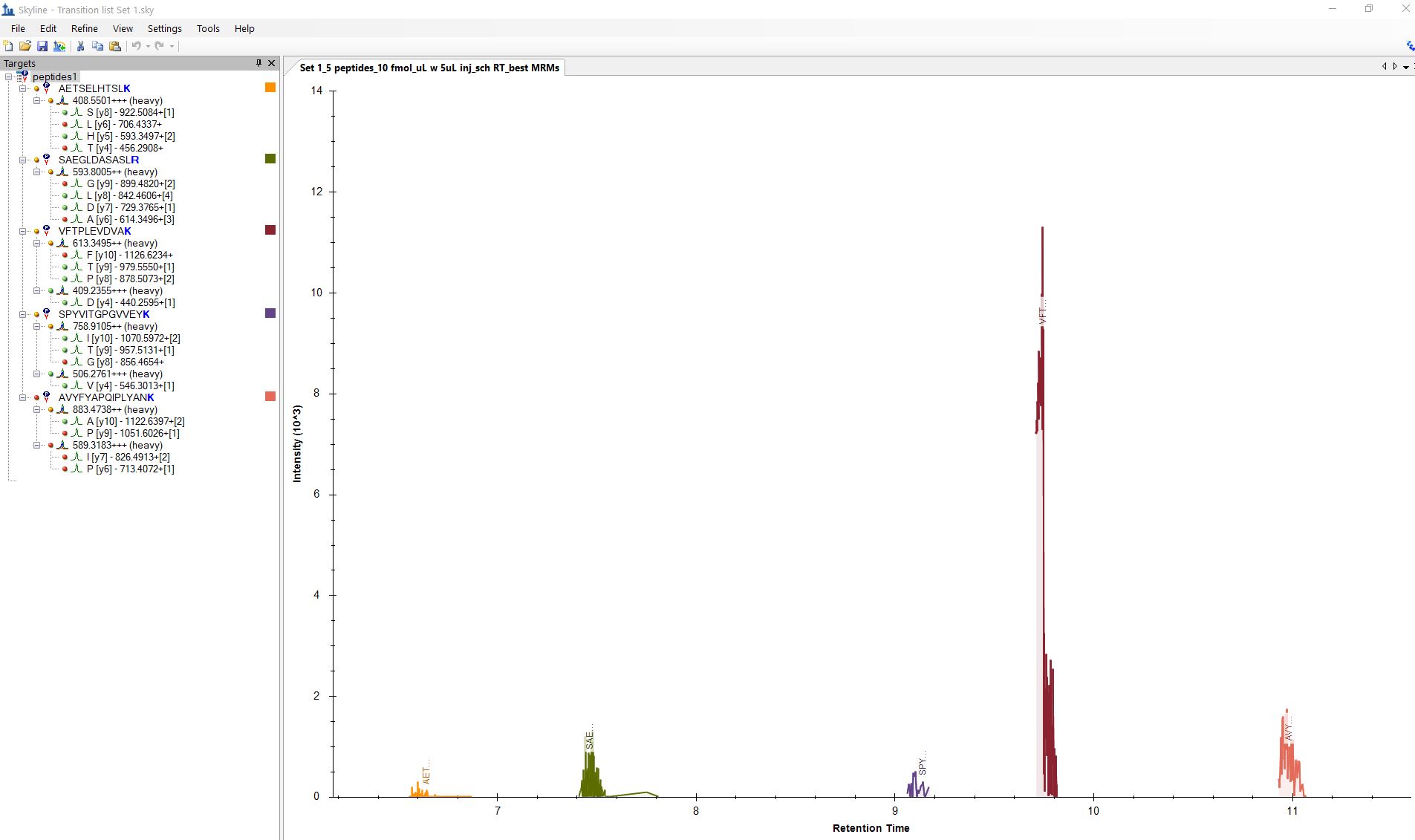 Screenshot of Data Import_peak issue.JPG
Screenshot of Data Import_peak issue.JPG