| Peak quantitation/ integration issue for high resolution small molecule data | chevans | 2021-10-26 11:36 | |||||||||
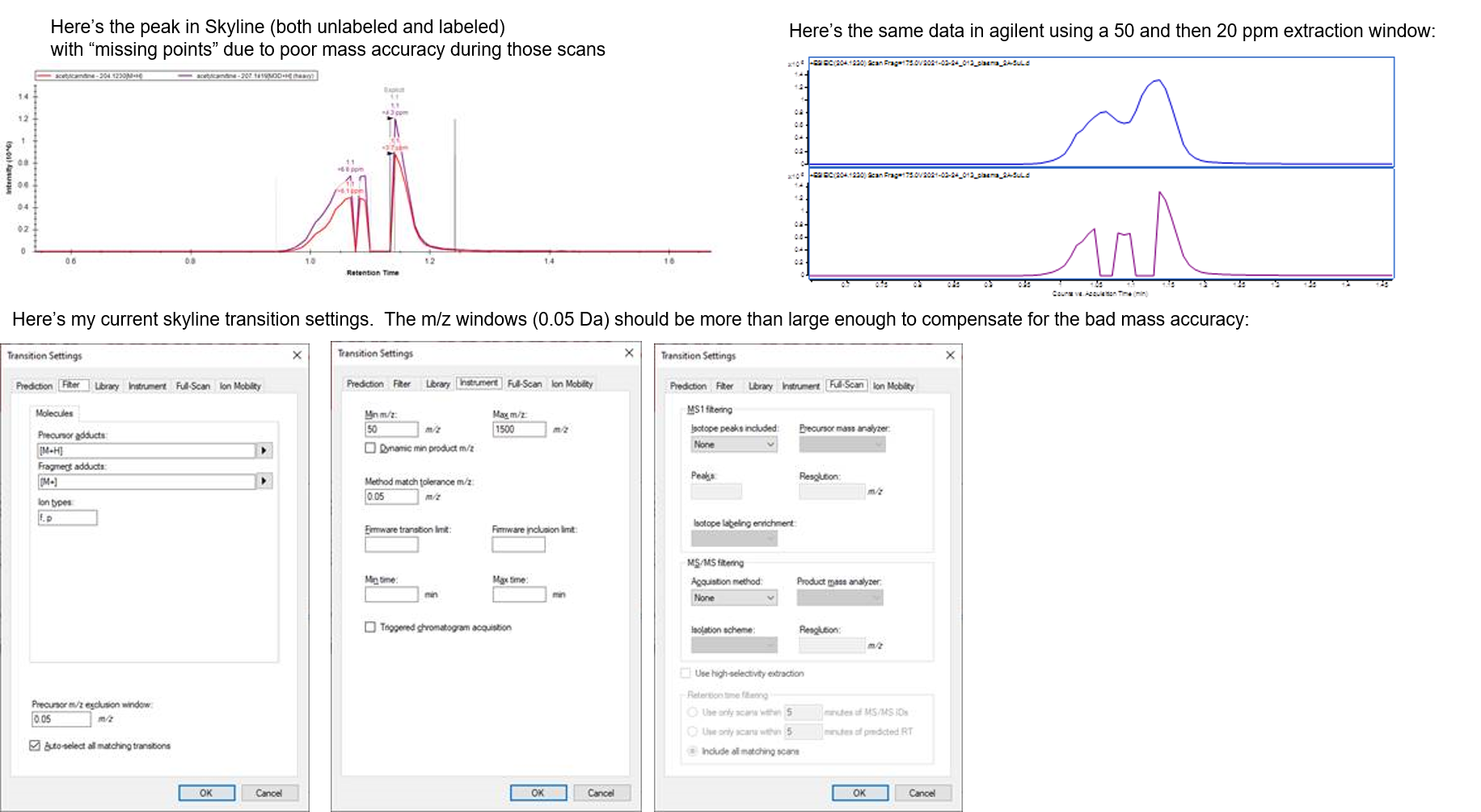
Apologies in advance for what may be an elementary question. I’m trying to use Skyline for analysis of small molecule data collected on an Agilent TOF. I have a data set in which, for one or a few data files, the mass accuracy wasn’t as good as it should have been for a few scans (probable intermittent loss of reference ion due to co-elution with salts or other interfering species). Those scans fall in the middle of a few peaks I’m trying to quantitate; for example, acetylcarnitine. This particular peak has an ugly "split" appearance due to poor retention on the column, but Skyline handles it fine for most of the data, the issue only crops up when the mass accuracy for a few scans is poor. In those cases, skyline is integrating only one part of the peak. I’d like to widen the “mass extraction window” (not the "skyline" for it, I realize, but concept should be the same) so I can get integrate the whole peak. This works fine in Agilent Quant software. But no matter what parameter I try changing in the “transitions settings” menu, the split peak doesn’t change and the integration is wrong. The attached image illustrates the problem. I could just use Agilent Quant for this project but I’m hoping to use it to analyze data on multiple different instruments and I like the “unifying” nature of Skyline software. Obviously an even better fix would be to solve the mass accuracy issue, but this crops up often enough that I'd like to be aware of a workaround, if one exists. Thanks very much for your help! Charles |
|||||||||||
| |||||||||||
 Skyline hi res quant question.png
Skyline hi res quant question.png