| |
| Brendan MacLean responded: |
2021-06-14 06:47 |
Please provide your system specs. This kind of extremely poor performance is usually due to insufficient memory. You can check this by running the Task Manager in performance mode and ensuring that memory consumption never reaches the maximum available. If it does, then processing will slow to a crawl as the system attempts to provide the require memory by swapping to disk.
We recommend at least 16 GB on your system for this type of processing and currently prefer 64 GB ourselves because those systems are now relatively inexpensive.
Check the system requirements page under Tips in the table of contents to the right of the installation page.
Look forward to hearing more of what you find regarding your system. Thanks for posting to the Skyline support board.
—Brendan
|
| |
| Brendan MacLean responded: |
2021-06-14 10:12 |
|
| |
| ref p responded: |
2021-06-14 20:07 |
Dear Brendan,
I'd love to thank for your instant and helpful response. I checked the memory occupation during chromatogram extraction, indeed, the Windows 7 was out of memory (99% occupied), specifically, you can move to Figure 1 for that information.
The specifications of my system are:
Windows 7, a 64-bit operating system;
Intel(R) Xeon(R) CPU E5-2667 v4 @ 3.2GHz;
Installed memory (RAM) 32.0 GB
According to the current available facility in my laboratory, the only possible way is to run Skyline computing on a Linux Server (Ubuntu 18.04.1 LTS). Is it executable?
|
|
| |
| Brendan MacLean responded: |
2021-06-14 21:25 |
That should be enough. How many transitions are there? Are you positive you have settings that keep Skyline from trying to extract all of your transitions over the entire gradient? How much parallelism are you allowing? I thought you said single file.
If you can use File > Share to save your entire project to a .sky.zip file and upload it to
https://skyline.ms/files.url
I can have a look at your settings and maybe figure out why it is consuming so much memory.
—Brendan
|
| |
| ref p responded: |
2021-06-14 22:47 |
Dear Brendan,
I have uploaded my Skyline document in *.zip format (v21.1) containing 2 replicates of example DIA thermo *.RAW data through the portal you provided. Please check and give me a response. Your are so kind for helping me a lot!
Uploaded files:
- iRtDIASupProt210606.sky.zip
- example Thermo Raw data.rar
Sincerely,
Guihua Jia
|
| |
| Nick Shulman responded: |
2021-06-14 23:37 |
Thank you for sending your Skyline document and those two .raw files.
On my computer, Skyline could extract chromatograms from those two raw files, and it took 15 minutes and Skyline never tried to use more than 20GB of RAM.
I cannot think of a reason that Skyline would be using significantly more memory on your computer.
You might want to try rebooting your computer and see if things speed up.
It might be helpful if you could send us a screenshot of the "Details" tab in Task Manager when Skyline is extracting chromatograms and has slowed to a crawl. You should sort on the "Memory" column so that the Task Manager grid displays the programs that are consuming the most memory at the top of the list.
Your Skyline document has 4 million transitions in it. Your Retention Time Filtering settings at "Settings > Transition Settings > Full Scan" tell Skyline to only extract chromatograms in a 5 minute range around the predicted iRT retention time. I would expect a 32GB computer would be able to handle a dataset of this size.
-- Nick |
| |
| ref p responded: |
2021-06-15 00:41 |
Dear Nick,
As what you said, a 15-min importing was also experienced myself at the very beginning. As the time goes by, Skyline slowed to a crawl at some point, but I missed it. I will provide the corresponding info. as you suggested.
Thanks a lot.
Guihua Jia |
| |
| ref p responded: |
2021-06-16 03:07 |
Dear Nick, Brendan,
Greetings.
Herein, I report the news:
1. I checked Skyline memory consumption status again as methods you suggested. Briefly, the computer was restarted and all the HDDs were cleaned up. **Skyline still runs quite slow**. Last night, this step, specifically, importing 10077 proteins containing 6 transitions per peptide (2MC, trypsin) considering 2+/3+/4+ precursor charge states and +1 b-/y-ions, took nearly 1.5 hours. During this period, chromatogram extraction was even not processed. **Skyline is the most memory consuming App.** (**Figure 2**), the total memory occupation reached the maximum 27% at the end of this stage, but it never exceed the previous reported 99% memory.
2. It was responded positively by Brendan when I suggested to try to run Skyline computing on an Ubuntu 18.04.1 LTS server (**Figure 3**, **May be it was clearly wrong, but I still need to confirm with you @ Brendan**). Unfortunately, at present, I did not find a Skyline installation software that may suitable for Linux server, including necessary documents. Please help me on this.
Sincerely,
Guihua Jia |
|
| |
| Nick Shulman responded: |
2021-06-16 07:28 |
Skyline only runs on Windows. The response "That should be enough", meant that the 32GB in your Windows 7 computer should be enough to extract chromatograms for your Skyline document with 4 million transitions.
I am not sure I understand what you are saying is slow now. When you said that chromatogram extraction took more than 20 hours, it sounded like the problem was that your computer did not have enough memory for everything that it was trying to do at once.
Skyline is written in the C# programming language. When Windows does not have enough RAM to hold everything that a C# program needs, things really do slow suddenly down by a factor of about a million. This has to do with the way that the garbage collection algorithm requires periodically looking at all of the pieces of memory that are in use.
What were you doing that took 90 minutes? That does not sound too bad. The steps that we would go through to troubleshoot something like that are different than what we would do for something that is taking more than a day.
-- Nick |
| |
| ref p responded: |
2021-06-16 09:23 |
Nick,
Thanks so much. I am quite clear at this time.
Guihua Jia |
| |
| Brendan MacLean responded: |
2021-06-16 11:43 |
Hi Guihua Jia,
I am sorry I didn't get to look at your files sooner. I have attached a PowerPoint slide deck with the changes I made to your file, which allowed me to run a complete analysis in around 6.5 minutes per file on my 5-year-old i7 with only 16 GB of RAM, on a spinning HD.
Your original document contained 4.9 transtions, which I felt was unnecessary. One key settings mistake was choosing "From filtered ion charges and types plus filtered product ions" (a setting I have considered removing). This setting subverts the "Pick [ 6 ] product ions" setting you used, since it asks Skyline to pick 6 transitions from the library, but then also to add "plus filtered product ions". Looking at the transitions in your precursors, you could see that there were more than 6 transitions at times and that some of them were not in the library spectra.
In my document, I also decided to go with 50% as many decoys as targets. This is valid, and I have shown in my own work that 25% as many decoys as targets works quite well with the Pan Human library from the Aebersold lab. If you want to stick with exactly the same number of decoys as targets, that should work well for you also, with your 32 GB of RAM.
I also switched to extraction from centroided spectra, because the MacCoss lab has found that preferable to using profile mode spectra with resolving power extraction. I am working on updating the Skyline basic DIA tutorial, which may have been the source of some of your problem settings, like using "last ion - 1" to filter your transitions. This was a mistake in that tutorial and several of our published experiments.
Thanks for sharing your files. I hope the attached explanation is helpful in getting you going on successful processing of your DIA data with Skyline.
--Brendan |
|
| |
| ref p responded: |
2021-06-20 02:59 |
Dear Brendan,
Many many thanks for providing informative suggestions on the Skyline document settings in the kind PPT file.
I even switched to extraction from centroided spectra under the recorded transition settings in Import Performance Fixes.pptx. Unexceptionally, Skyline still keeps going to a crawl, as I observed, when more than 8 DIA files (similar than the previously provided .RAW file in ''example Thermo Raw data') have been imported before. Namely, when the 9th DIA data was importing, the Skyline started running slower and slower (Figure 1, When the Skyline goes to a crawl).
Fortunately, I noted that, on average, the importing time is about 10 min per DIA raw, however, the most time consuming stage is Joining File after the completion of importing. At this Joining file stage, the memory cosumption is the biggest, and the Skyline running speed is the slowest.
Eventually, I have successfully imported all of my results (~25 GB) within 30 minutes after resorting to Skyline computing on a PowerEdge R740xd Windows Server, and the average importing time significantly reduced to 1 minute. I also found that the Joining file still costs the most of the memory (Figure 2, Memory consum. PowerEdge R740xd).
Hopefully, what I have provied here may become a source of thinking for improvement.
Best regards,
Guihua Jia
|
|
| |
| Nick Shulman responded: |
2021-06-21 23:58 |
I am not sure that Skyline really is slowing to a crawl at the end of the chromatogram extraction. I think it might just be that there is a lot of work that needs to be done that is not reflected in a change in the "Importing Results..." status graph.
When you have an iRT predictor, Skyline reads through the entire raw file twice. The first time through the raw file, Skyline extracts chromatograms for only the iRT standard peptides. This is why the first 40% of the progress bar passes quickly. Then, things seem to stop for a bit while Skyline is figuring out over what time range the rest of the peptides should have their chromatograms extracted. The second pass through the raw file goes much slower than the first pass, because Skyline is extracting chromatograms for many more peptides. After all of the spectra have been read from the raw file the second time, Skyline still has a lot of work to do in terms of detecting peaks in the chromatogram.
When Skyline is all done with peak detection, the "Importing Results..." status graph disappears. Skyline still has to do two things:
1. Joining the skyd files. When Skyline extracts chromatograms from more than one raw file, the chromatograms are first put into a separate .skyd file for each raw file. When chromatograms for all of the raw files have been extracted, a single large .skyd file is created which contains the chromatograms from the separate .skyd files.
2. Updating results. Skyline goes through all of the peptides in the document and sets their peak areas etc. to reflect the values obtained from the new chromatograms.
These two last steps happen at the same time, but only one of those get to appear on the status bar. So, when the status bar says "Joining file Xxx", I'm not 100% sure whether the joining is really taking that long, or whether it's really the updating results.
I am not sure whether it's a cause for concern that Task Manager shows Skyline using a lot of memory. If no other programs need the memory, the Microsoft .Net Runtime will happily delay performing garbage collection, and use more memory than is strictly necessary. Things do not really slow to a crawl until it is trying to use more memory than is available, and no amount of garbage collection is able to reduce that number below the amount of RAM in the computer. When that happens the disk usage spikes up a lot as memory gets written to disk and then immediately needs to be read again. I am not sure exactly how you are supposed to recognize that in Task Manager. I think it might involve looking at "PF Delta", or maybe just disk usage.
When you saw that joining was taking a lot of memory, had you told Skyline to import 9 result files at once? Or had you asked Skyline to import 8 result files, allowed that to completely finish, and then told Skyline to import one more result file?
In the first case, Skyline would be joining nine small .skyd files together.
In the second case, Skyline would be joining one large .skyd file with a single small .skyd file.
I will make sure that Skyline is not using a undue amount of memory for this joining process, but I need to know what exactly you had asked Skyline to do.
Thanks for all the info.
-- Nick |
| |
| Brendan MacLean responded: |
2021-06-22 08:47 |
My take on Guihua Jia's last response is a bit different (please correct, if I am missing something):
- I think you are saying that you are able to import everything in 30 minutes (perhaps using some parallelism) after switching to a PowerEdge R740xd with 64 GB of RAM (per Task Manager) and for processors "Up to two 2nd Generation Intel® Xeon® Scalable processors, up to 28 cores per processor". Under which Skyline would default to importing all 9 files in parallel with "Many" chosen.
- Importing each single DIA raw data file on your i7 with 32 GB of RAM takes on average 10 minutes. (Presumably importing 1 at a time?)
- On your i7 with 32 GB of RAM the computer again ends up thrashing (consuming all memory and attempting to swap to disk) during the Joining step.
This last one makes perfect sense, given that you are showing the operation taking close to 64 GB of RAM on the PowerEdge machine. Looking at the memory consumed on a high-memory computer is a good way to get a sense of what it might require on a lower memory computer.
It might be interesting for us to get all of your files (.sky.zip and 9 raw data files) to reproduce this issue exactly. Also, as Nick indicates, it would help to have a screenshot of how you are choosing to import the raw data files. I assume you are always trying to import all 9 at once but possibly changing your choice of "Files to import simultaneously" (One at a time, Several, Many).
One more thing to consider is that using Skyline Batch (https://skyline.ms/batch.url) allows you to perform this import with the Skyline command-line interface, which will always consume less memory because it does not need to maintain the user interface and undo stack. I don't think that is likely to solve a memory consumption issue with the joining step, but it might be worth a try to see what the difference looks like.
Thanks for all your time and feedback. I can think of work we have not done yet, which would decrease memory consumption during the joining phase. Perhaps your case will prove to be the motivation we need.
--Brendan
|
| |
|
|
 memory consumption.svg
memory consumption.svg Figure 2-Skyline memory consu.svg
Figure 2-Skyline memory consu.svg Figure3-Brendan's response.JPG
Figure3-Brendan's response.JPG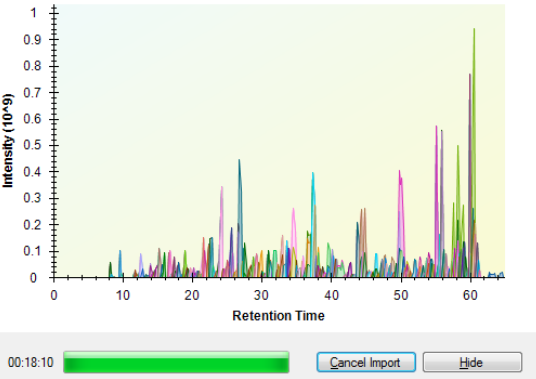 When the Skyline goes to a crawl.PNG
When the Skyline goes to a crawl.PNG Memory consum. PowerEdge R740xd.JPG
Memory consum. PowerEdge R740xd.JPG