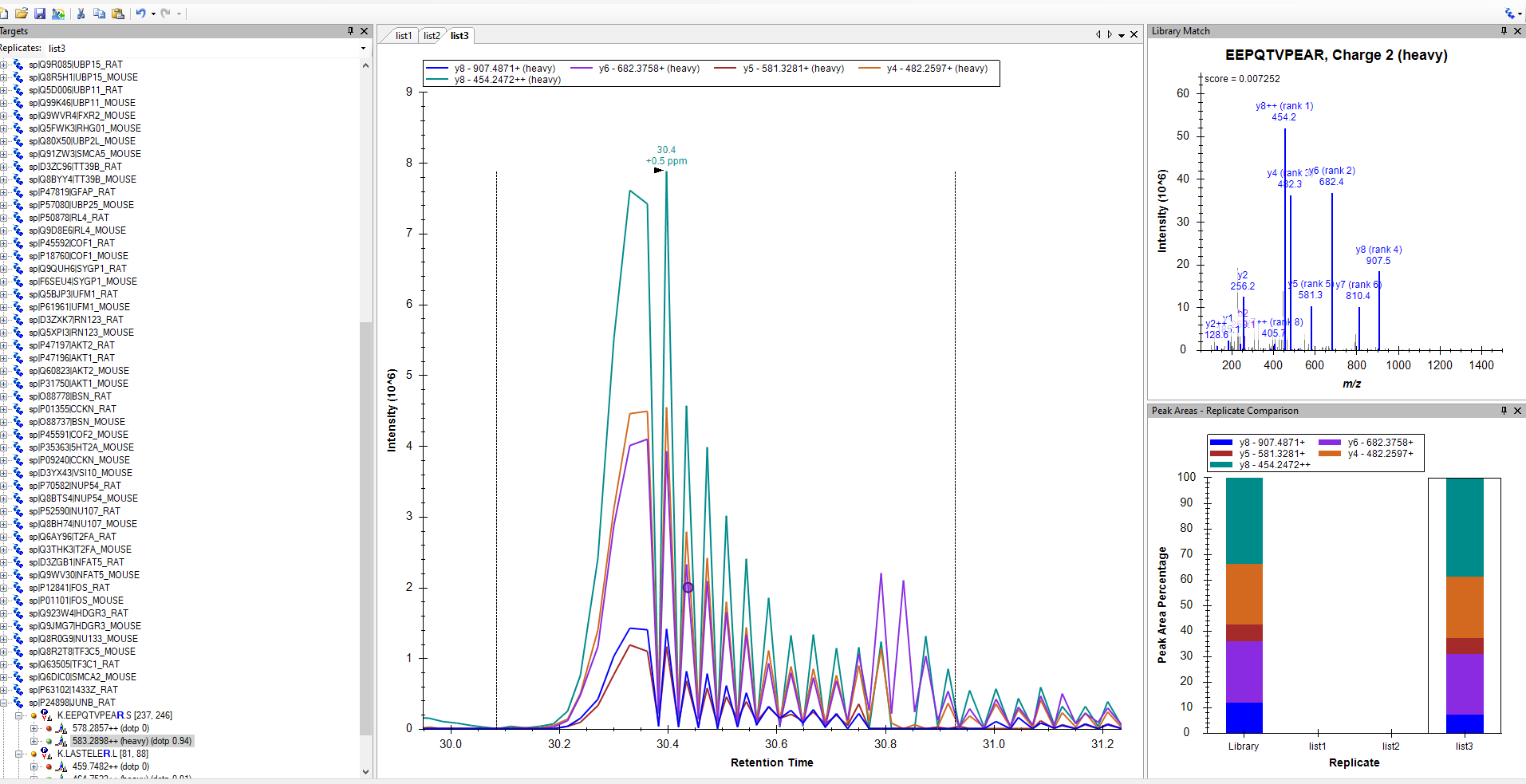
Chromatograms that look like that are an indication that two different sorts of MS2 spectra contributed to the chromatogram.
One of the easiest ways to diagnose what is going wrong is to click on the chromatogram. When you click on the chromatogram it brings up the Full Scan viewer which shows you what the spectrum that contributed to that point on the chromatogram looked like. You can push the Next and Previous buttons on the Full Scan viewer and see what is different about the spectra that yielded a high intensity compared to the spectra that yielded a low intensity.
What do you have specified as your acquisition method and isolation scheme at "Settings > Transition Settings > Full Scan"?
If your acquisition method is "DIA", then there are few things that might cause the chromatograms to look like that.
A common reason for this happening is that your isolation windows overlap by a small amount (maybe one m/z unit).
That is, maybe you have one isolation window that goes from 574-584, and you have another isolation window which goes from 583-593, and the reason that you did this is because you know your mass spectrometer filters out a lot of ions near the edge of the isolation window.
If you are using the isolation scheme called "Results Only", Skyline will assume that a precursor whose m/z is 583.2898 can be found in both of those isolation windows.
What you should do instead is change your isolation scheme to the one called "Results (0.5 margin)". Then, Skyline will interpret those isolation windows as being 574.5-583.5 and 583.5-592.5, and will only think that 583.2898 can be found in the first isolation window.
Another reason that Skyline might be making chromatograms looking like that is that you have a custom isolation scheme which does not actually match the isolation windows in your raw file. If you have a list of Prespecified Isolation Windows in the Edit Isolation Scheme dialog, make sure that they actually match what is in your raw file.
A third reason that chromatograms sometimes look like that is that you asked the mass spectrometer to collect some completely other type of scan. Sometimes people tell the mass spectrometer to alternate between ETD and HCD, and one of those things results in very poor fragmentation. In that case, I would recommend that you use ProteoWizard msconvert to create a .mzML file where the spectra that you don't want Skyline to see have been removed.
Hope this helps.
If you still can't figure out what is going wrong, you can send us your Skyline document and one of your raw files.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file and your raw file are less than 50MB you can attach them to this support request.
Otherwise, you can upload them here:
https://skyline.ms/files.url
-- Nick
 1.png
1.png 2_heavyvslight.png
2_heavyvslight.png