Abdullah,
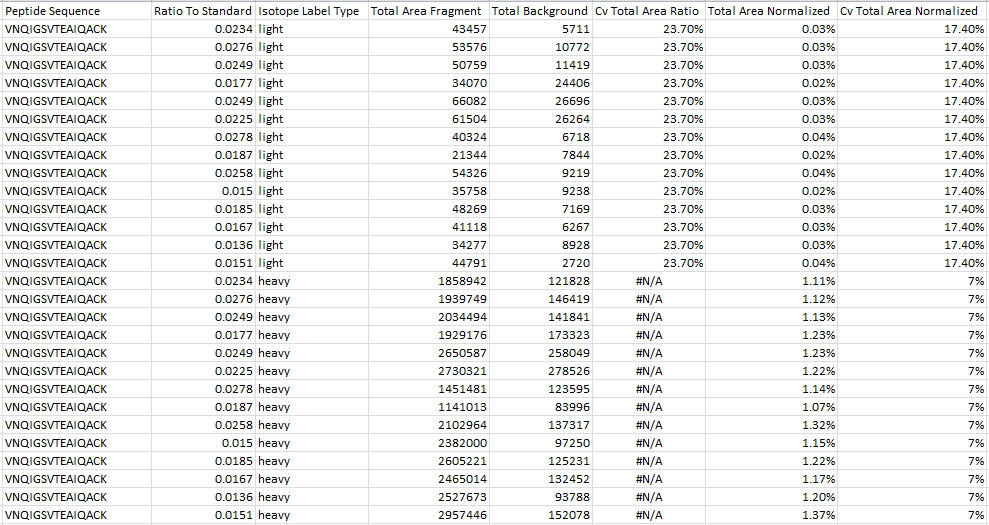
We do not recommend that you use the column called "TotalAreaNormalized". That column was added to Skyline a long time ago before we anticipated all of the different ways that users might want to normalize their results. If you hover over the column header in the Document Grid you can see that the tooltip says:
The TotalArea normalized to the sum of the TotalArea values
for all peptide precursors in the document
The column that we do recommend that you use is called "Normalized Area" and it's located under:
Proteins > Peptides > Peptide Results > Quantification > Normalized Area
This is the area normalized according to whatever normalization method you have specified in the Peptide Settings dialog at:
Settings > Peptide Settings > Quantification
It sounds like you should choose the normalization method "Ratio to Heavy".
I am not sure what could be causing the two separate peaks that have so many transitions in common. One thing that can happen to your sample in the test tube is deamidation where a couple of different amino acids sometimes undergo a transformation which results in a net mass loss of 1 Dalton. This chemical change has a small effect on the retention time of the peptide. Also, because it is such a small mass change, the new precursor usually falls in the same DIA isolation windown, and many of the same transition masses will be observed.
-- Nick |
 Skyline exported data.PNG
Skyline exported data.PNG image.png
image.png