| |
| Brian Pratt responded: |
2019-05-16 10:14 |
Hi Fabian,
Can you provide a copy of the transition list you're using? Also the Skyline document you're using (File > Share > Complete) would be useful to understand your current Skyline settings. You can upload to http://skyline.ms/files.url or whatever is convenient for you.
Thanks for using the Skyline support board!
Brian Pratt
|
| |
| Fabian responded: |
2019-05-17 03:11 |
Dear Brian,
I did so, I named the excel and the sky file:
Isotopes via Insert Small Molecule Transition List.
Btw. Assuming there is no miss at my side, having such a column as the insert option could solve the issue?
Fragment Ion
precursor
precursor [M+1]
precursor [M+2]
Kind regards
Fabian
|
| |
| Brian Pratt responded: |
2019-05-20 13:44 |
Hi Fabian,
The easiest way to do this is to let Skyline calculate the isotopes for you. Just include each precursor once:
Trypsin pep_VATVSLPR C37H67N11O11 [M+2H]
Trypsin pep_VATVSLPR C37H67N11O11 [M+2NH4]
Trypsin pep_VATVSLPR C37H67N11O11 [M+2Na]
Trypsin pep_VATVSLPR C37H67N11O11 [M+H+NH4]
Trypsin pep_VATVSLPR C37H67N11O11 [M+H+Na]
then go to Settings > Transition Settings > Filter and make sure "Auto-select all matching transitions" is checked, then go to the Full-Scan tab and set "Isotope Peaks Included" to "Count" and set "Peaks" to 3.
That will give you a set of transitions like this:
Transition, Precursor, Product Charge, Product Mz, Product Ion Formula, Product Neutral Formula, Product Adduct
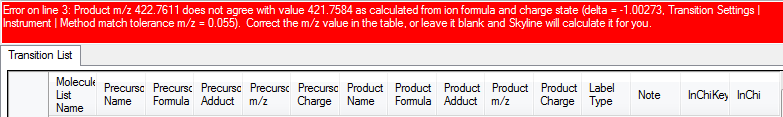
precursor[M+2H], 421.7584[M+2H], 2, 421.758352, C37H67N11O11, C37H67N11O11, [M+2H]
precursor[M+2H] [M+1], 421.7584[M+2H], 2, 422.259787, C37H67N11O11, C37H67N11O11, [M+2H]
precursor[M+2H] [M+2], 421.7584[M+2H], 2, 422.761082, C37H67N11O11, C37H67N11O11, [M+2H]
precursor[M+2NH4], 438.7849[M+2NH4], 2, 438.784902, C37H67N11O11, C37H67N11O11, [M+2NH4]
precursor[M+2NH4] [M+1], 438.7849[M+2NH4], 2, 439.286295, C37H67N11O11, C37H67N11O11, [M+2NH4]
precursor[M+2NH4] [M+2], 438.7849[M+2NH4], 2, 439.787566, C37H67N11O11, C37H67N11O11, [M+2NH4]
precursor[M+2Na], 443.7403[M+2Na], 2, 443.740297, C37H67N11O11, C37H67N11O11, [M+2Na]
precursor[M+2Na] [M+1], 443.7403[M+2Na], 2, 444.241731, C37H67N11O11, C37H67N11O11, [M+2Na]
precursor[M+2Na] [M+2], 443.7403[M+2Na], 2, 444.743026, C37H67N11O11, C37H67N11O11, [M+2Na]
precursor[M+H+NH4], 430.2716[M+H+NH4], 2, 430.271627, C37H67N11O11, C37H67N11O11, [M+H+NH4]
precursor[M+H+NH4] [M+1], 430.2716[M+H+NH4], 2, 430.773041, C37H67N11O11, C37H67N11O11, [M+H+NH4]
precursor[M+H+NH4] [M+2], 430.2716[M+H+NH4], 2, 431.274325, C37H67N11O11, C37H67N11O11, [M+H+NH4]
precursor[M+H+Na], 432.7493[M+H+Na], 2, 432.749325, C37H67N11O11, C37H67N11O11, [M+H+Na]
precursor[M+H+Na] [M+1], 432.7493[M+H+Na], 2, 433.25076, C37H67N11O11, C37H67N11O11, [M+H+Na]
precursor[M+H+Na] [M+2], 432.7493[M+H+Na], 2, 433.752055, C37H67N11O11, C37H67N11O11, [M+H+Na]
Thanks for using the Skyline support board!
Brian Pratt
|
| |
| Fabian responded: |
2019-05-21 06:45 |
Hey Brian,
thank you for your response!
Unfortunately, that is not the way which works for me.
The reasons are the following:
In that case skyline extracts for all my targets 3 peaks (mass traces) from the raw file.
Consider a very small molecule (with a peptide like isotope distribution), in that case the +2 Isotopic peak will not be observable so that extracting the +2 Isotope will increase interference and possibly decrease correct peak assignement.
In another case I might have an isotopic distribution so that the +4 Isotope is very abundant, so it is very helpful to extract the mass trace of it.
Therefore I build a skyline file which has optimal individual isotope selection - optimized per molecule.
What is missing is an option to transfer this list via the "Insert Small Molecule Transition List" option.
Kind regards
Fabian
|
| |
| Brian Pratt responded: |
2019-05-21 08:56 |
Hi Fabian,
I see what you mean. This isn't something that's come up before. You could of course manually disable the unwanted isotopes but that is not scalable.
What would be your ideal format for expressing your wishes to Skyline? A new column for "Isotope"? Or to have Skyline notice the seemingly conflicting m/z value and work out for itself that it's an isotope?
Thanks
Brian
|
| |
| Brendan MacLean responded: |
2019-05-21 09:34 |
You can also choose the "Percent" option for "Isotope peaks included" and then specify a "Min % of base peak", which may behave more like what you are seeking, with more peaks included for larger molecules. Even when you choose just "Count" and give a straight "Peaks" value, Skyline will not include anything with less than 1% of the overall expected isotope distribution. So, very small molecules may still not see a +2 isotope transition.
Not necessarily suggesting we shouldn't also consider enabling what you are requesting, but as it is not immediately working, I thought I should explain fully the existing options for controlling isotope peak inclusion. It is not as simple as just including a specific count no matter what.
|
| |
| Brendan MacLean responded: |
2019-05-21 09:35 |
What algorithm are you using to choose the isotope peaks in your list?
|
| |
| Fabian responded: |
2019-05-22 03:07 |
Dear Brian and Brendan,
thank you both for your replies!
@Brian: I think, having something like "Fragment Ion" as one has it in the Document Grid could solve this. Of course, if skyline guess the isotopes based on conflicting m/z values that would also be cool. Also I am not sure if that could cause troubles in other scenarios?
@Brendan: thanks for giving me the insights in the details. For me this solution has a few drawbacks, for the following reasons:
I want to make sure that I can import a transition list e.g. from an excel sheet exactly in the way it is constructed in the excel, without an skyline interpretation based on rules (1% isotope, %...), also this skyline rules are of course meaningful in most cases.
Besides keeping the original unmodified "excel" in skyline, there are mainly two reasons related to this: First, for certain sample types it can be that a certain isotope regularly cause interference - better having it excluded, a simple solution is to not use the mass trace of it at all.
Second, for unknown molecules where the chemical structure is mainly used as an hypothesis and to ease the screening of the mass of interest in skyline, in such case the isotope distribution could be totally different than expected from the chemical formula, because the formula only fits to the mass but not to the real formula. In that case skyline can not make educated guesses and therefore it would be great to have an option to circumvent it.
Kind regards
Fabian
P.S. For initial peak recognition I use the internal peak determination of skyline without using prophet or so. In cases this is not sufficient I set peak boundaries.
|
| |
| Brendan MacLean responded: |
2019-05-22 08:31 |
Hi Fabian,
Thanks for your feedback. I can't really imagine Skyline becoming quite as flexible as you appear to be seeking in the near-term. I don't think we even allow precursor isotopes on molecules where we don't know the chemical formula, and we don't have a way of supporting users specifying what the isotope distribution should be or certainly the exact m/z for +1 or +2, etc.
I think the best Brian can offer in any kind of near-term release would be to allow you to select among the isotopes Skyline automatically calculates from the chemical formula, which will have their own caclulated m/z (no matter what you put in your Excel spreadsheet) and relative abundance as part of an isotope distribution, and they would be restricted to only the set of peaks with at least 1% of the entire isotope distribution. We limit the calculations at that point no matter what.
Because people and the ad hoc tools they write often produce mistakes, giving Skyline a transition list is a negotiation, where the provider of the transition list specifies what is trying to be measured and Skyline ensures that this somehow matches the known constraints of atomic masses. Most of the time this protects researchers from a lot of errors. And Skyline always uses its own calculated values when a chemical formula is present.
For small molecules, we have increased the flexibility a great deal, allowing straight m/z specification for the monoisotopic m/z and fragment ions, but we have not yet gone that far for isotopic distributions, and it is not yet a priority for us to do that. It will still take more people looking for that functionality to raise its priority for us.
In the meantime, we will see what it would cost to allow transition list import to allow the arbitrary specification of which isotopic peaks we know about are targeted and which are not. Would this be enough for you?
I am still curious about how you are coming up with your list of precursor isotopes, and your chemical formulae. Are they really just designed to produce the desired mass without knowing they are actually correct? Chemical formulae will be necessary for us to have any kind of isotope distribution, but still our idotp (Isotope Dot Product) score depends on the expected distribution of the atomic isotopes. More information on what you are doing might help to convince us this is something that should be enabled for you and others.
Thanks again for explaining your intended experiment.
--Brendan
|
| |
|
|
 Insert_Transition_List.PNG
Insert_Transition_List.PNG