| Large scale PRM data (msx count= 8) | Wael | 2019-03-20 09:01 | |||||||||||||||||||||||||||||
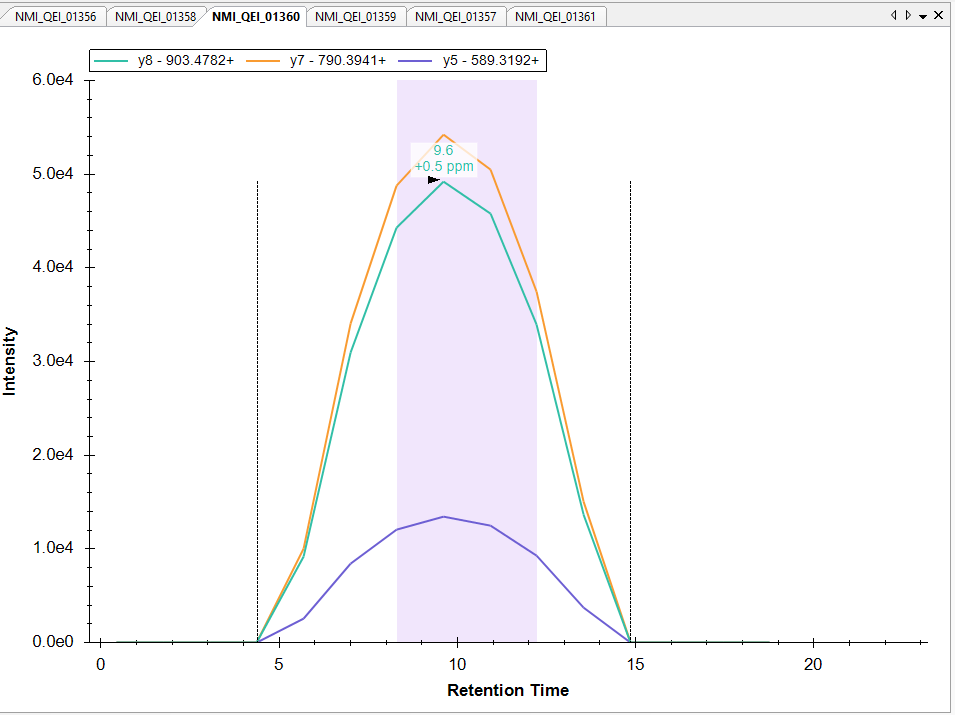
Dear Skyline team, Is skyline able to process such a workflow and if yes how can I improve the peak integration, pelase ? Please let me know if you would like me to share skyline file with you. Many thanks |
|||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
 Large scale PRM data.PNG
Large scale PRM data.PNG