Now we want to put some theoretical library on the skyline, however,
there are some issues with the application.
The theoretical library record each peptide with relative intensities
which are all no greater than 1. In the theoretical library, we put
the following information, for example, given this peptide:
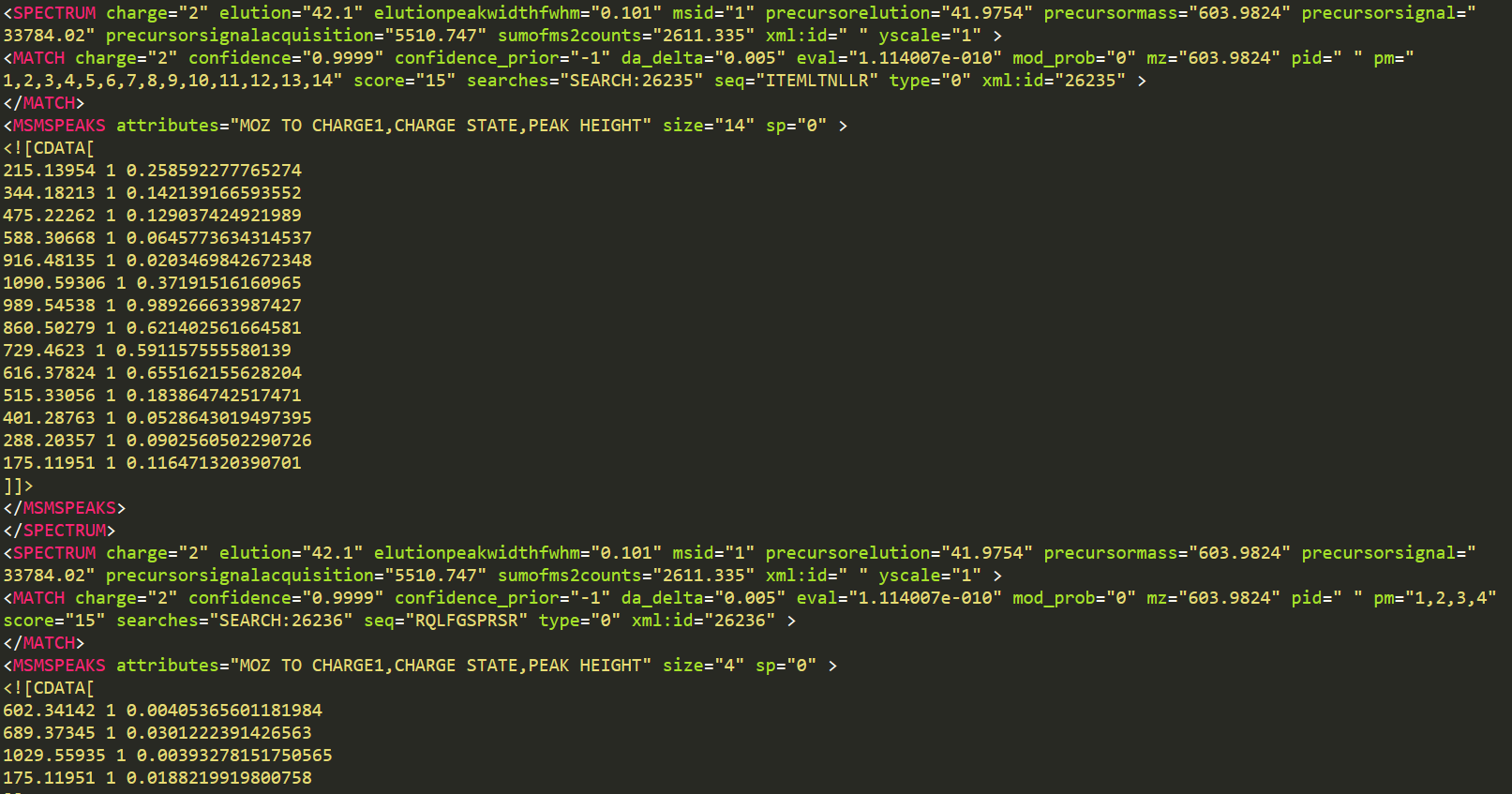
<SPECTRUM charge="2" elution="56.82013" elutionpeakwidthfwhm="0.153"
msid="1" precursorelution="56.846" precursormass="766.4388"
precursorsignal="2379.753" precursorsignalacquisition="2085.837"
sumofms2counts="2514.072" xml:id=" " yscale="1" >
<MATCH charge="2" confidence="0.9999" confidence_prior="-1"
da_delta="0.005" eval="1.114007e-010" mod_prob="0" mz="766.4388" pid=" "
pm="1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16" score="15"
searches="SEARCH:34641" seq="ELYPIALPFLDIK" type="0" xml:id="34641" >
</MATCH>
<MSMSPEAKS attributes="MOZ TO CHARGE1,CHARGE STATE,PEAK HEIGHT"
size="16" sp="0" >
<![CDATA[
243.13445 1 0.342857450246811
406.19778 1 0.0905757695436478
503.25054 1 0.0276164971292019
616.3346 1 0.00763198733329773
687.37171 1 0.0444362200796604
800.45577 1 0.0330323725938797
1289.75091 1 0.0753663927316666
1126.68758 1 0.983879029750824
1029.63482 1 0.122389301657677
916.55076 1 0.46008637547493
845.51365 1 0.207167357206345
732.42959 1 0.577380180358887
488.30842 1 0.0218868106603622
375.22436 1 0.0373408868908882
260.19742 1 0.0576274879276752
147.11336 1 0.0267378911375999
]]>
</MSMSPEAKS>
</SPECTRUM>
The issues are:
1, The retention time cannot be aligned correctly which only showed a
narrow range from around 60~80 min which should be from 3~80 min;
2, The ranked peak included "y1" fragment ions for most peptides with
theoretical library and some of the peak areas graph cannot be shown.
also, what is "sumofms2counts" in <SPECTRUM>? Could you please help me
to figure out what is the problem?
Thank you very much! |
| |
| Nick Shulman responded: |
2019-03-04 06:20 |
I am not sure I understand your question.
Are you saying that you have some Protein Pilot search results, and there is something wrong with the spectral library that Skyline (actually BiblioSpec) builds from that?
Or are you writing a program that produces .group.xml files, and you want to know what data to put in the file so that Skyline will be able to read it?
If you send us your .group.xml, we can take a look at it.
If your files are less than 50MB, you can attach them to this support request.
Otherwise, you can upload files here:
https://skyline.ms/files.url
-- Nick |
| |
| sunbergsoon responded: |
2019-03-04 06:30 |
Hi Nick,
You are right, we wrote a new ".group.xml" file in which are all the theoretical spectrums, we just want to use the minimum information to edit it and to
define the theoretical library so that Skyline will be able to read it. I will put a sample into that link.
Thank you very much for your kind help.
Best regards,
Sunberg |
| |
| Nick Shulman responded: |
2019-03-04 12:33 |
Thanks for sending that file.
The problem is that all of your spectra have the same xml:id attribute (they are all " ").
For this reason, BiblioSpec gets confused when it tries to retrieve a particular spectrum, and so every spectrum in the library ends up looking exactly the same.
If you gave each of those spectra a different id, eg xml:id="1", xml:id="2", etc., then I think everything would work.
-- Nick |
| |
| sunbergsoon responded: |
2019-03-04 23:53 |
Hi Nick,
It did work!
Thank you so much for your help!
Another, what is "sumofms2counts" in <SPECTRUM>? Does this parameter matter for BiblioSpec to select the right precursor ions and how to calculate it?
Best regards,
Sunberg |
| |
| Nick Shulman responded: |
2019-03-05 11:04 |
BiblioSpec does not pay attention to the sumofms2counts attribute. I don't know what that attribute would be supposed to be equal to.
The mzs and intensities of the spectrum get stored in BLOBs in the RefSpectraPeaks table of the .blib file. (.blib files are SQLite databases).
I'm not sure what you're asking about BiblioSpec selecting the right precursor ions. BiblioSpec sets the precursorMz and precursorCharge in the RefSpectra table based on attributes in your <Spectrum> and <Match> table.
BiblioSpec does not know anything about peptide fragmentation or product ions.
Skyline reads the spectrum from the RefSpectraPeaks table. Skyline generates all of the fragment ions based on the users settings in:
Settings > Transition Settings > Filter
and using the "Ion match tolerance" on:
Settings > Transition Settings > Library
and Skyline figures out which peptide transitions had the highest intensity in the library spectrum.
-- Nick |
| |
| sunbergsoon responded: |
2019-03-06 04:01 |
Hi Nick,
Thanks a lot for these days' help and I really appreciate it.
Also, thank you for your kind explanation and I see.
With my best regards,
Sunberg |
| |
|
|
 skyT.PNG
skyT.PNG