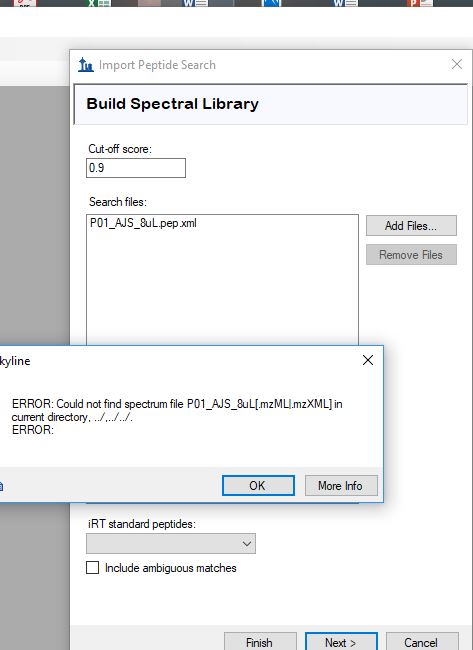
Skyline uses another program called "BiblioSpec" to read peptide search results. BiblioSpec does not know how to read the same file formats as Skyline does. For this reason, it is best to put your .mzML files in the same folder as your .pepXML files. (We recommend using .mzML instead of .mzXML, since .mzML supports chromatograms, but both formats should work).
What do you mean that it fails when it tries to use the mzXML as the result file? Can you tell us what error you're seeing?
I am not sure what your other question is. Are you saying that you did "File > Import > FASTA", and Skyline gave you all of the tryptic peptides from the FASTA file, instead of the ones that were in your .pepXML files?
You can tell Skyline to only give you the peptides that are in the spectral library (which was built from the .pepXML files). That settings is at:
Settings > Peptide Settings > Library
If you have a library in your document, then there is a dropdown there "Pick peptides matching", and make sure that it is set to either "Library" or "Library and Filter", so that Skyline will only give you peptides that are in your library.
Another way to add library peptides to your document is to go to:
View > Spectral Libraries
and push the "Add All" button.
If you are adding peptides from the spectral library viewer, it is recommended that you first set up a background proteome by going to:
Settings > Peptide Settings > Digestion
A background proteome will enable Skyline to associate peptides with proteins.
By the way, the "Could not find spectrum" error message is poorly worded. It is using the "|" character to mean "or". It is saying that it was not able to find either "P01_AJS_8uL.mzML" or "P01_AJS_8uL.mzXML", and that it tried looking in the current directory, the parent of the current directory, and the parent of the parent of the current directory.
-- Nick |
 skyline.JPG
skyline.JPG