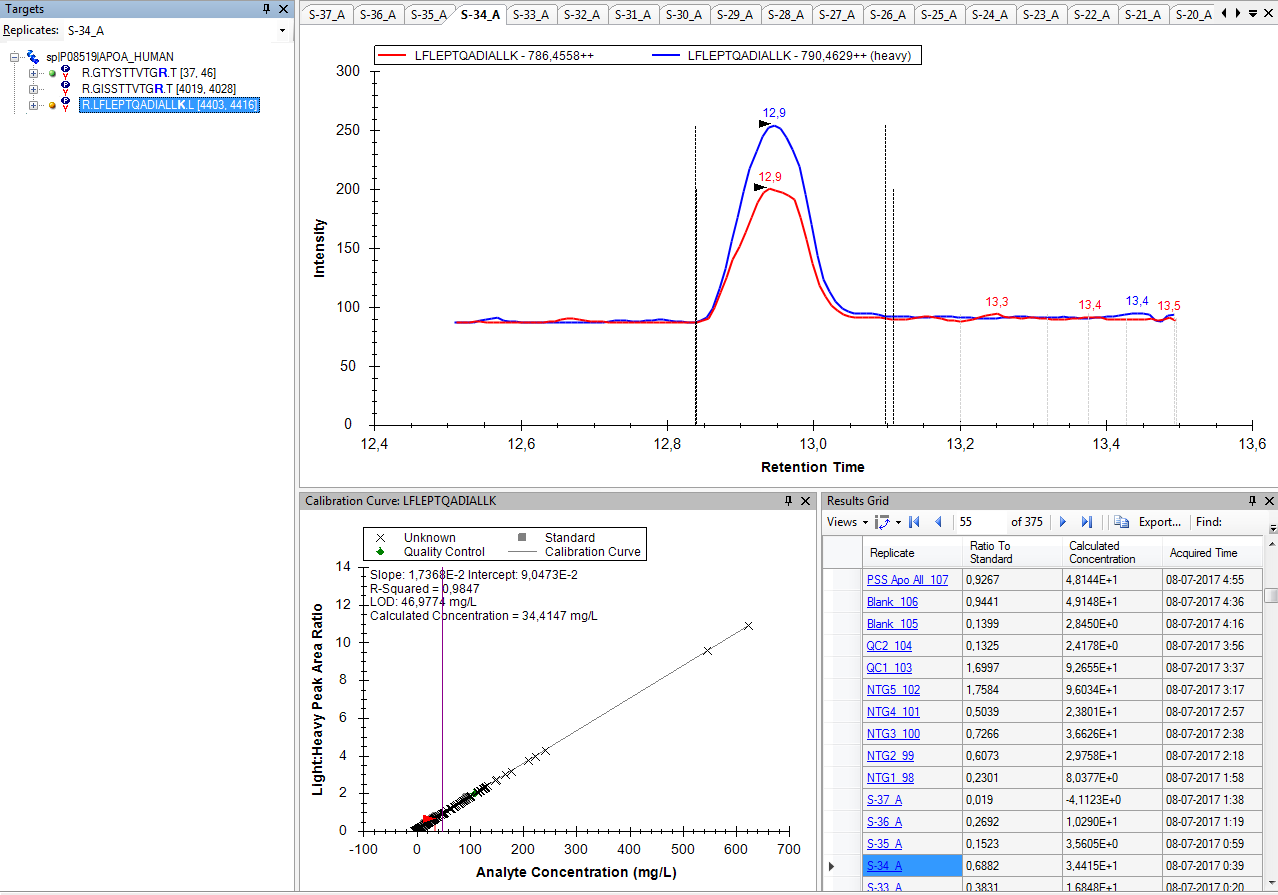
I do not know what happened, but, for some reason, Skyline chose not to integrate the heavy form of that peptide.
That is, if you look at the chromatograms for the heavy transitions of that peptide, there are not vertical dotted lines indicating the location of the chosen peak.
You can tell Skyline to do the peak detection again by going to:
Edit > Manage Results > Rescore
and if you do that, then Skyline will choose reasonable peak boundaries for all of your peptides.
I am not sure how things got this way, or whether this is a bug that we need to fix. I could imagine that something like this could happen if you added the heavy peptide to your Skyline document after you had already imported chromatograms from the raw files. Usually whenever you add new transitions to your Skyline document you need to do "Edit > Manage Results > Reimport" in order to see chromatograms for the new things. However, for SRM data, Skyline can show you the new chromatograms immediately, but I can imagine that there might be quirky peak detection behavior like this if you don't do at least a rescore after that.
If you think your heavy and light peptides were already in your Skyline document when you did the "Import Results", then I should probably try to figure out what went wrong. It would be helpful if you could zip up one of your raw files (e.g. "NTG5_102.d") and send it to us.
-- Nick |
 Skyline no NaN.png
Skyline no NaN.png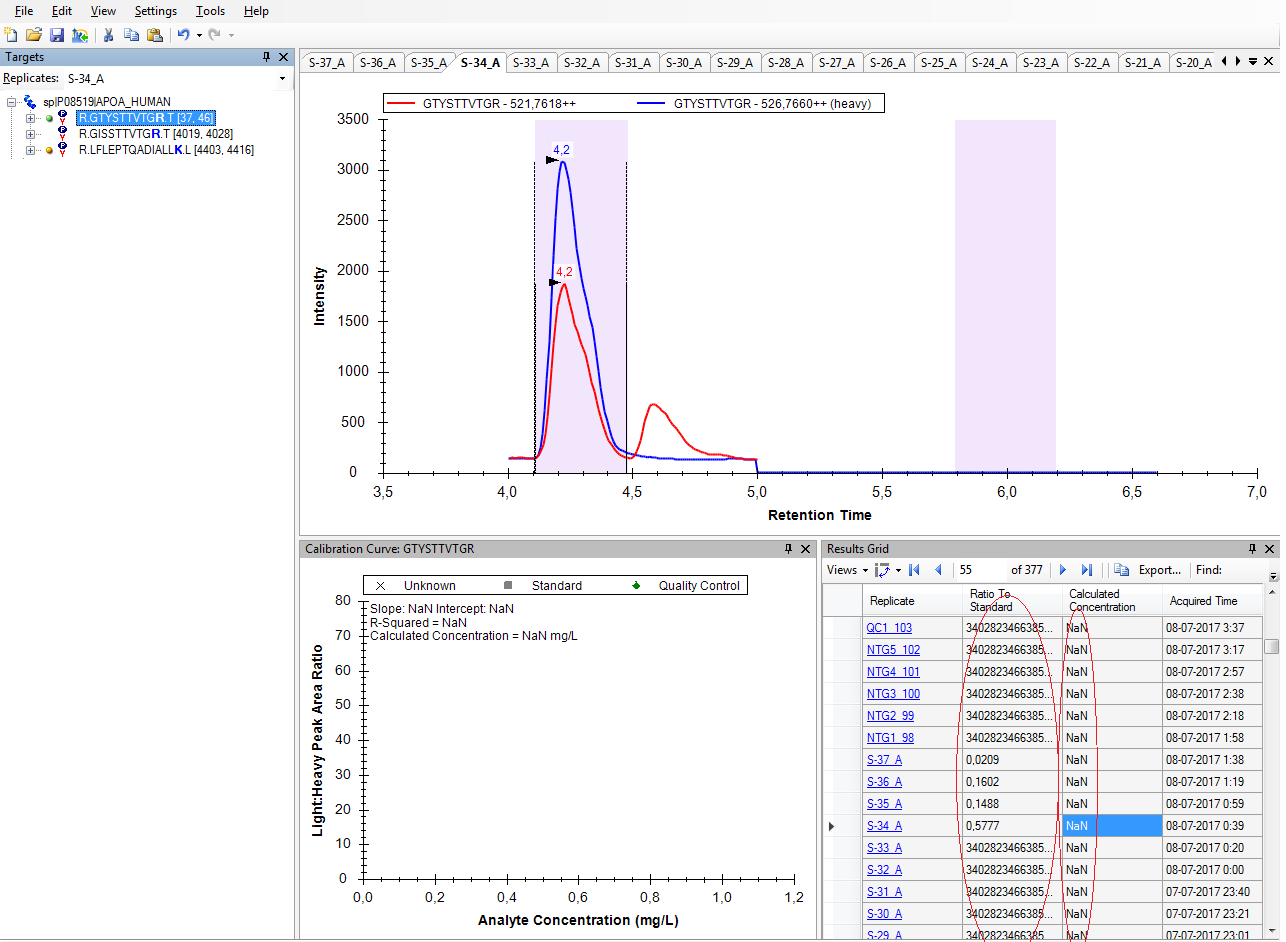 Skyline NaN.png
Skyline NaN.png