| Feature request: Monitoring [M-1] ion for small molecules | Chris Ashwood | 2018-08-08 07:42 | |||||||||||||||||||||||||||||||||||||||||
Hi Skyline Team, I'm working in the small molecule space and have noticed that sometimes, when using a wide extraction window (30ppm), my "new" targets are off-by-one errors of existing targets (e.g the monoisotopic peak for one target is within 10ppm of the first C13 isotope of another small molecule). This occurs due to my library-building DDA run performing MS2 on the 1st C13 isotope, and despite attempts at correcting this using Proteowizard's precursorRefine filter, there are still some uncorrected precursor masses that could mistaken for real targets. In this case, the ability to integrate or include the [M-1] ion in peak integration would provide a way to quickly evaluate the off-by-one error with the peak area window in Skyline, rather than inspecting the target in full-scan view for each suspect peak. I've observed that idotp with a count of 3 is not a sufficient parameter for mitigating the off-by-one error (0.91 dotp value for both the target and off-by-one target, attached picture). I don't know if anyone else would value this feature, as this issue could be significantly reduced at the hardware/method level by running at higher-resolution or centroiding the data prior to analysis with Skyline but thought I would suggest it, in case it was seen to be a useful feature. Cheers, |
|||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||
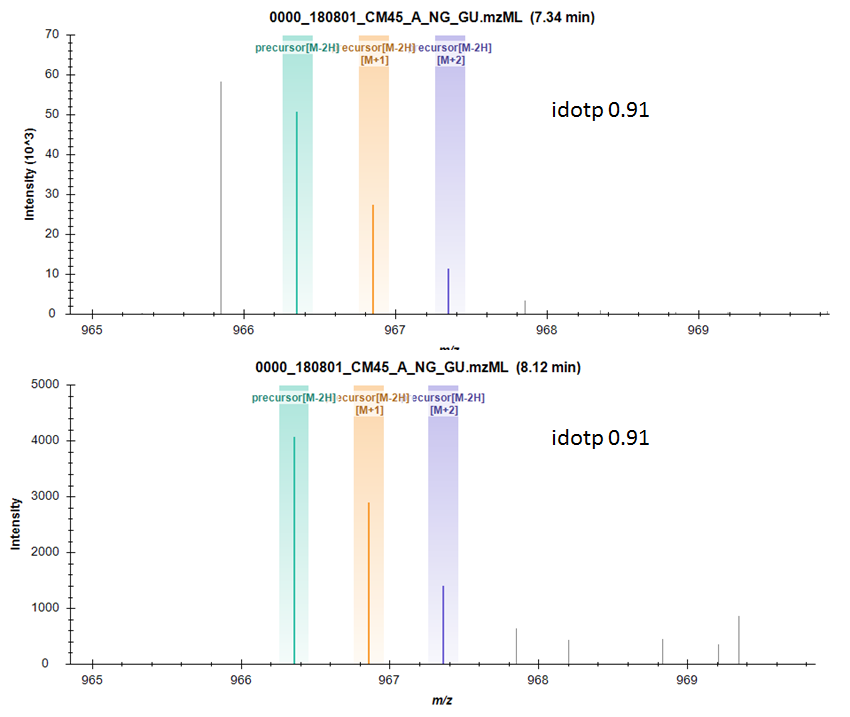 off-by-one.png
off-by-one.png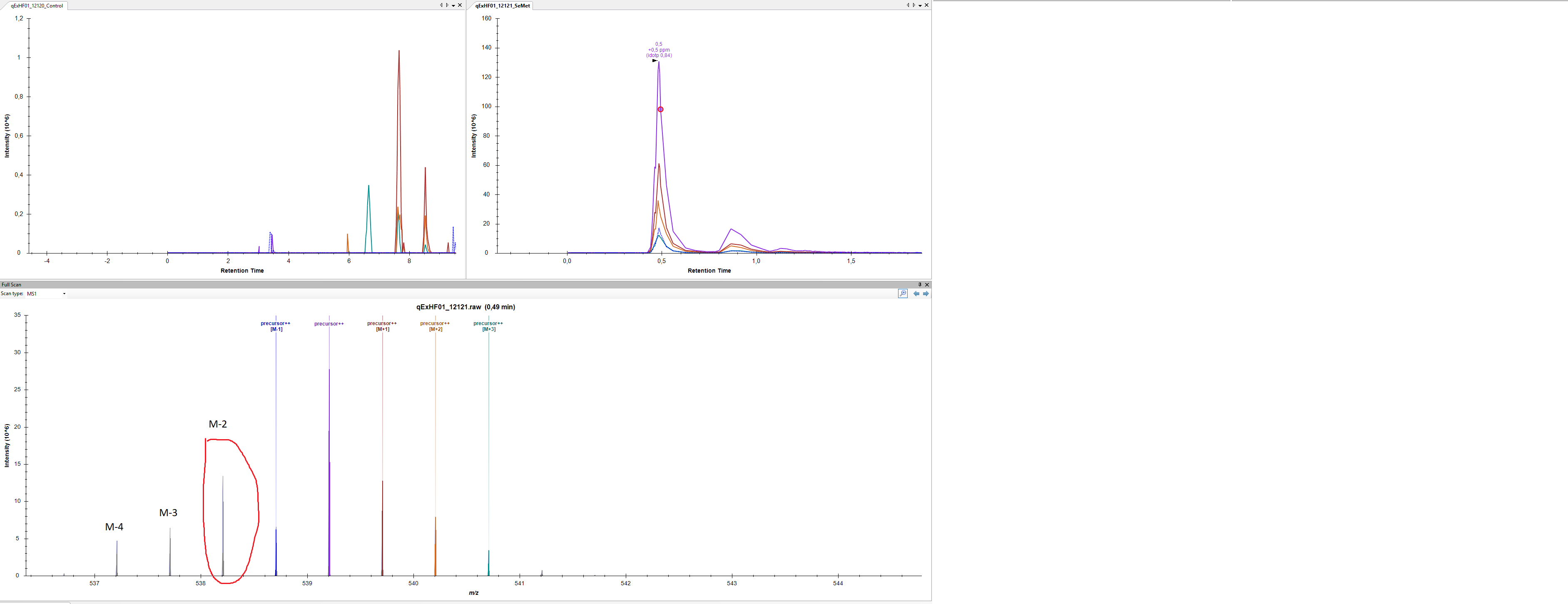 SeMet.PNG
SeMet.PNG