Hi Skyline team!
We are currently trying to set up Skyline to jump start from DDA to DIA data analysis ;)
We are using a Q-Exactive plus for this.
We have run a DDA experiment in order to extract information (RT, product ion etc...) from 38 proteins we are interested in and created a library based on maxquant andromeda search engine, using the msms.txt file.
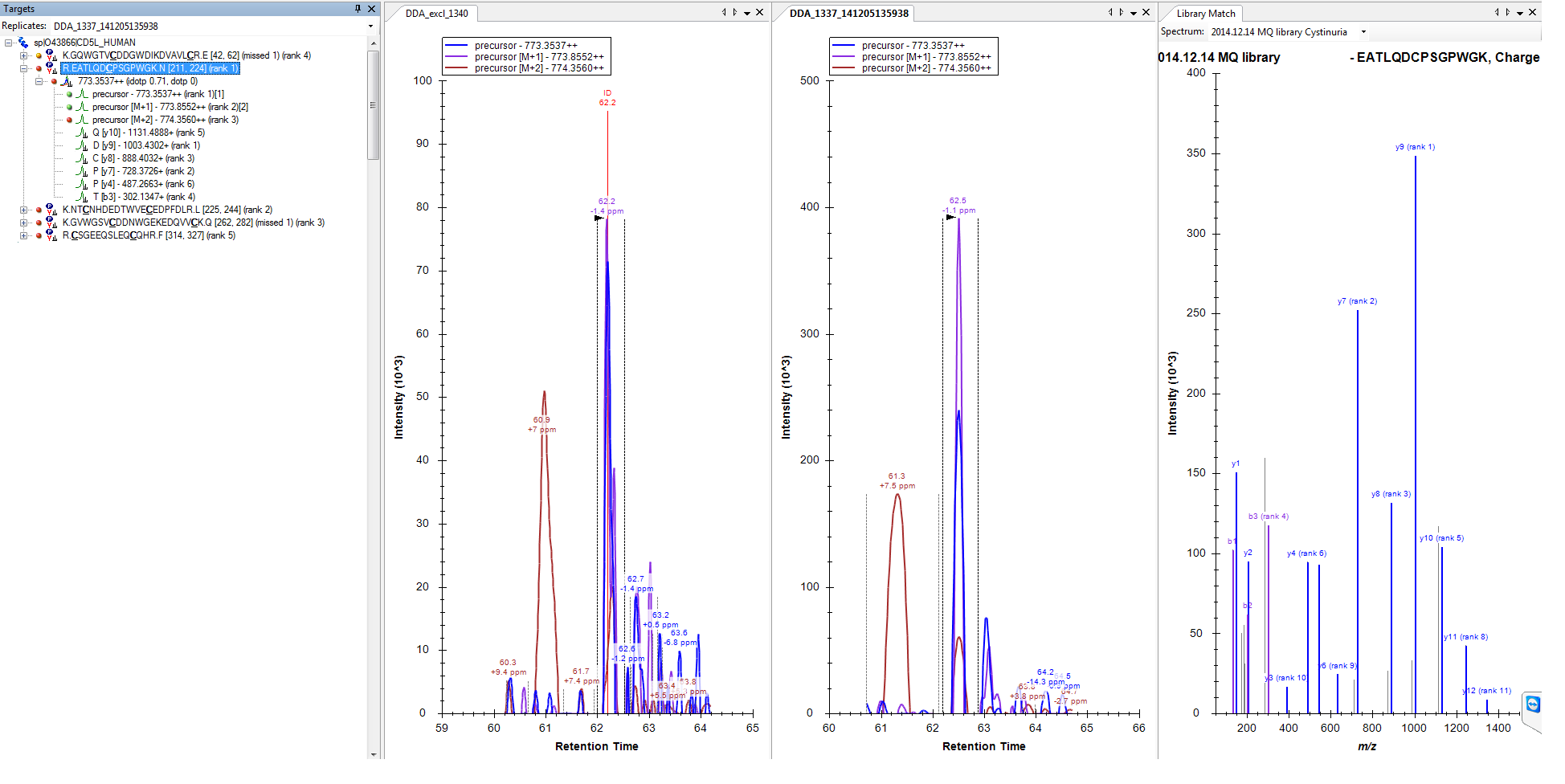
However, when I import the same raw file that was used for library building, there is a bunch of product ion that appears missing from the raw file...which is a nonsense to my mind, while the precursors are quite okay.
Do you have a guess on what could be happening?
Thanks a lot for developping such a nice software and for the support!
Matthieu |
| |
| Brendan MacLean responded: |
2014-12-14 09:53 |
Hi Matthieu,
It is sort of "nonsense" to be trying to extract fragment ion chromatograms from DDA data. I seriously considered covering this in Webinar #1, because so many people seem to want to try it, but we just didn't have the time. It is not possible to extract chromatogram from DDA MS/MS spectra, because they are not collected systematically over time for the same precursor m/z ranges, but instead semi-randomly based on the most intense MS1 signal at any given time, likely with some dynamic exclusion applied to keep from selecting the same m/z multiple times in close succession.
This means that if you get any fragment chromatograms at all, you should expect them to look like a single point spike fairly frequently, but only in runs where you also have a spectrum collected for the particular precursor m/z of interest.
Once you understand this, it won't be at all surprising that a run that lacks an ID annotation for your peptide also lacks any fragment ion chromatograms. It would be surprising if the run with the ID annoations (which is not active in your screenshot) lacked fragment chromatograms, but not surprising if it had only a single-point spike for fragment chromatograms.
Of course, if you do not have your MS/MS extraction parameters set up to perform extraction (which would be the recommended stat for DDA), then it is not surprising that you get no fragment chromatograms at all (which would be the normal expectation for DDA).
Please post a screenshot of your Transition Settings - Full-Scan tab.
I hope this helps to clarify. I will certainly consider adding a Skyline Tip on extraction of fragment ion chromatograms from the MS/MS scans in DDA data, complete with pictures.
Thanks for posting your request to the support board.
--Brendan |
| |
| Math responded: |
2014-12-14 11:43 |
Thanks for the quick answer, there are some point that seems more clear to me.
1) Normally the parameters to extract MS/MS are quite ok (see attached file 1), as for some precursors/product ion it was nicely extracted (2nd file attached).
2) And on the other hand I have proteins like on my original post, where there is nothing found (file attached 3, I have activated the run with the ID annotation this time).
3) And so I get that MS2 can be a single point spike, maybe that could explain some of my results? (attached file 4).
4) Lastly I have sometimes quite strange product ion extraction which I have some difficulties to explain (5th attached file). Or maybe it's a MS method problem...
Anyway thanks again for your time to answer our question! And I hope I am clear enough!
Matthieu |
|
| |
| Brendan MacLean responded: |
2014-12-14 12:11 |
The new images definitely add new information.
1. At least they look like I would expect to see from DDA, nothing at all like you would want to see for quantifiable chromatograms.
2. The only thing I can think of is that somehow the spectrum getting identified as your peptide of interest did not have an associated precursor m/z value that fell within "Method match tolerance m/z" (on the Transition Settings - Instrument tab) of the predicted m/z for this peptide. This could happen for a number of reasons, on a high accuracy instrument the most likely case would be that the instrument chose to isolated the M+1 peak instead of the monoisotopic peak. Whatever the reason, this means that the imported data did not have a spectrum with precursor m/z within your match tolerance.
3. Yes, this is the cause of all strange looking results for fragment ion chromatograms.
4. Again caused by the fact that you probably only have one point for most of your fragment ion chromatograms. Skyline is left to find some way of interpolating all other points based on the interval chosen from the MS1 scan times. It is not so noteworthy that sometimes Skyline just extends your one point to the extent of your chromatogram time range. There is not really a well defined behavior here, and I actually wonder more how Skyline figures out to bring the intensities back down to zero at some point, rather than that it sometimes fails to do so. With a single point it is not at all clear how intensity should be treated.
Thanks for posting the extra screenshots and questions.
--Brendan |
| |
| Math responded: |
2014-12-14 14:20 |
Thanks (again and again :) ) for all these answers, it is really helping me understanding, even the basics of DDA.
But now, our problem is that we wanted to select the best product ion on these DDA analysis, and then to go on targeted MS/MS methods, with the idea to develop absolute quantification on these.
If I understand well, now that we have the information on RT, we should start targeted tests in order to validate which transitions have a nice chromatogram and so are more suitable for the rest of our experiments?
Looking forward to your answer. |
| |
| Brendan MacLean responded: |
2014-12-14 20:36 |
|
| |
| Math responded: |
2015-02-03 08:23 |
Hi Brendan,
After a few weeks and some recent advances we made, I come back to you with a question.
We managed to see, in t-MS2, most of the peptides we are interested in, and with at least 3 transitions each.
However, we feel like there is an issue in our Q-exactive method.
On the attached picture, you can see the same precursors and fragments, in PRM and DIA, left and right, respectively.
(Don't mind the RT, they were not verified before MS analysis).
In DIA, the chromatogram seems smooth without any refinement, while the t-MS2 looks like a wave.
For us it looks like if there was to many measurement in t-MS2 and we don't have the optimum signal at each measurement.
Would you have a guess on what could be happening in here?
Oh and thank you for the webinars, they are very useful to us ;).
Have a nice day.
Matthieu |
|
| |
| Brendan MacLean responded: |
2015-02-05 12:00 |
Hi Matthieu,
This is normal. Your DIA method is measuring much fewer points across the peak, and therefore, naturally ends up with a smoother peak because it doesn't have enough points to actually capture the variance in the intensity measurements. There is a nice explanation of the impact of number of points across a peak in Lange et al., 2008, Mol Syst Biol.
I will just say that I remember a discussion in the early days of Skyline among CPTAC instrument operators, where someone showed a slide much like the image you have attached comparing unscheduled SRM with scheduled SRM. The person said essentially that he was used to seeing curves like the ones you have for DIA, but with scheduled SRM he often saw curves much more like the ones you have for PRM, and that he had learned that this was okay, and just the effect of having more point across the peak.
If you can achieve smoother peaks with the same number of points across them, that is, of course, better, but more points across the peak will simply more accurately measure the variance in your measurements, and decreasing the number of measurements across your peaks is just hiding that variance from you, not making it go away.
In the end, the peak areas from Skyline should be fine either way, as long as you have around 8 point across your peaks.
Thanks for posting this question. I am sure the discussion will be useful to others.
--Brendan |
| |
|
|
 2014.12.14 Skyline question.png
2014.12.14 Skyline question.png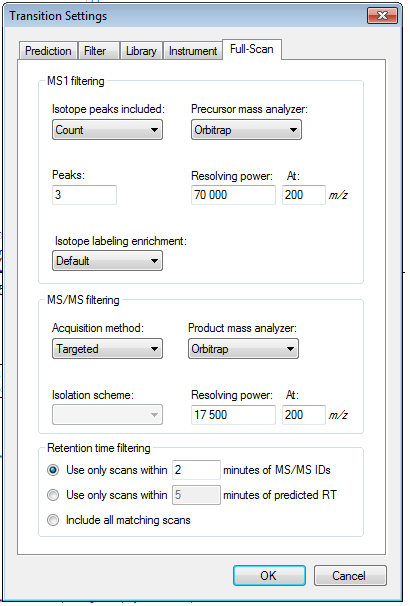 2014.12.14 Full scan parameters.png
2014.12.14 Full scan parameters.png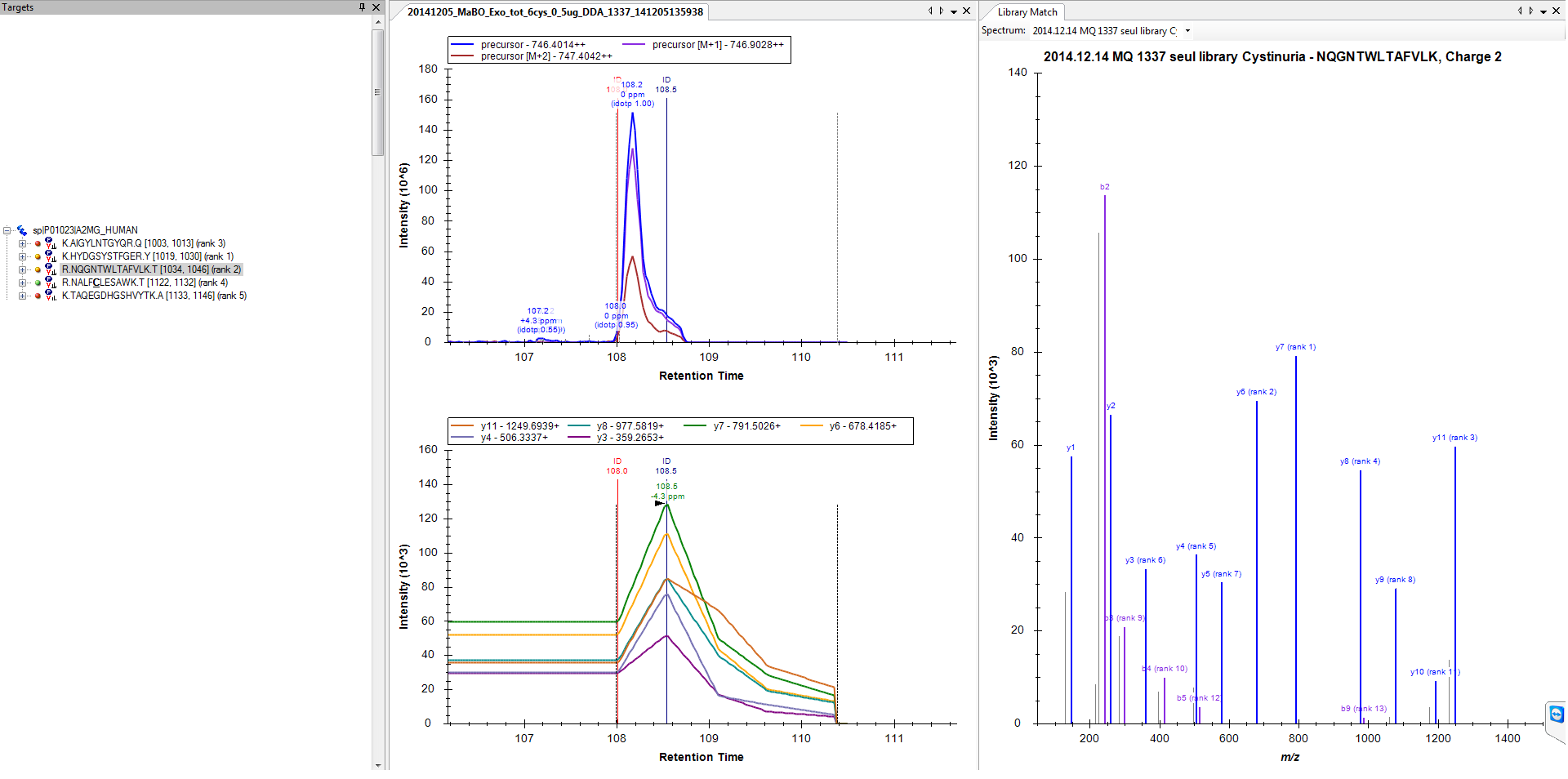 2014.12.14 nice extracted MS2.png
2014.12.14 nice extracted MS2.png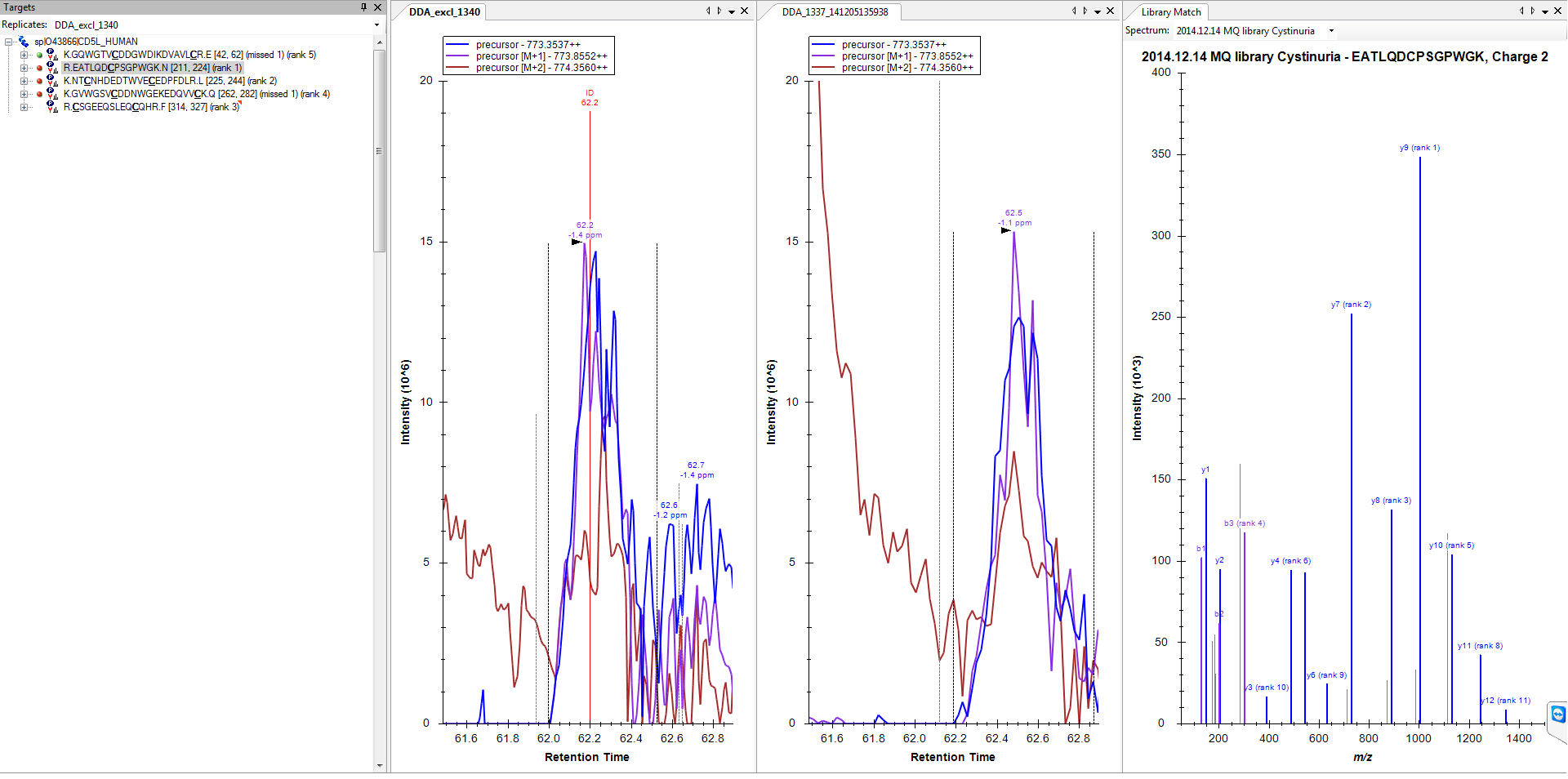 2014.12.14 Skyline question2.png
2014.12.14 Skyline question2.png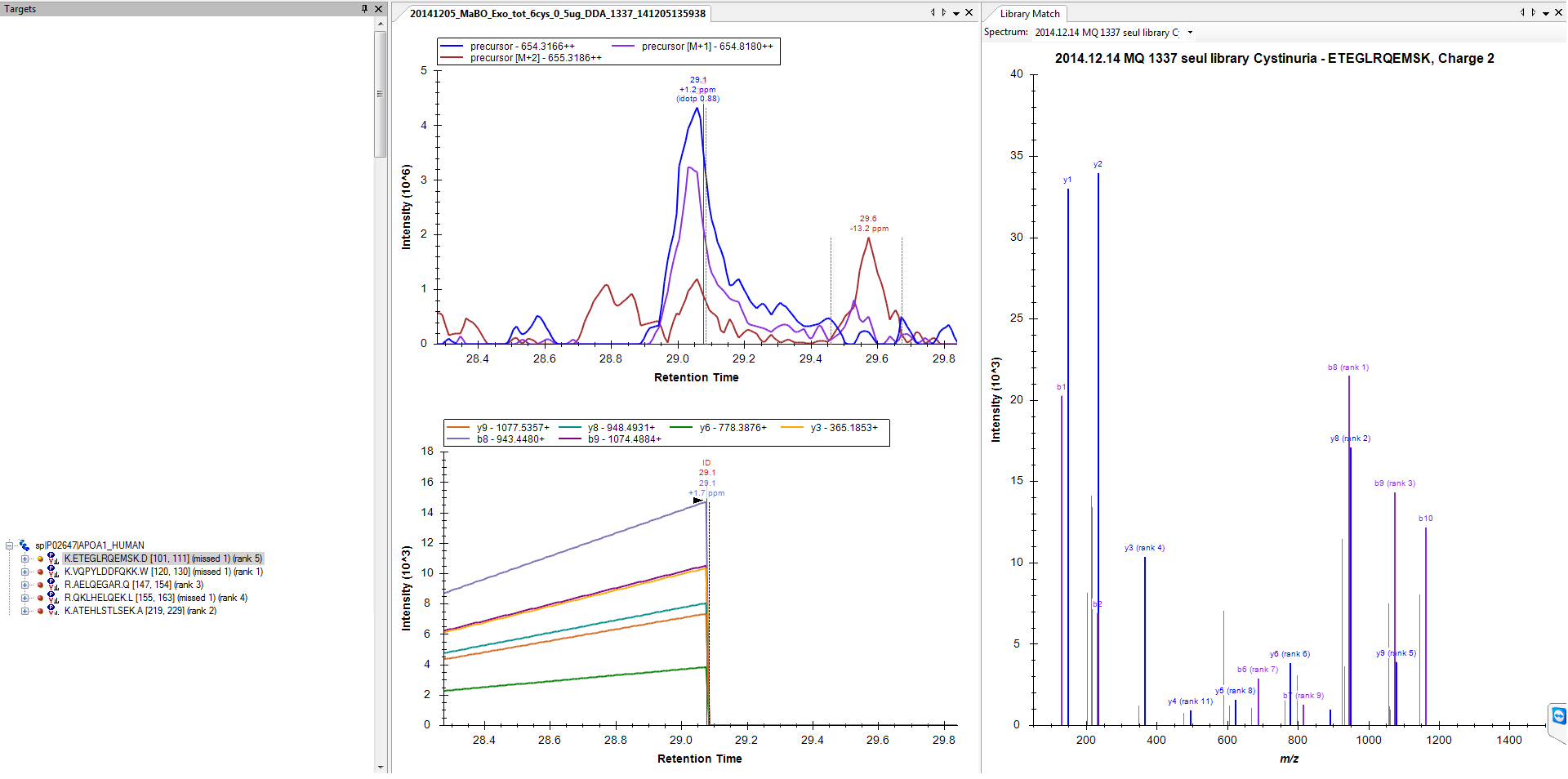 2014.12.14 ID single point spike.png
2014.12.14 ID single point spike.png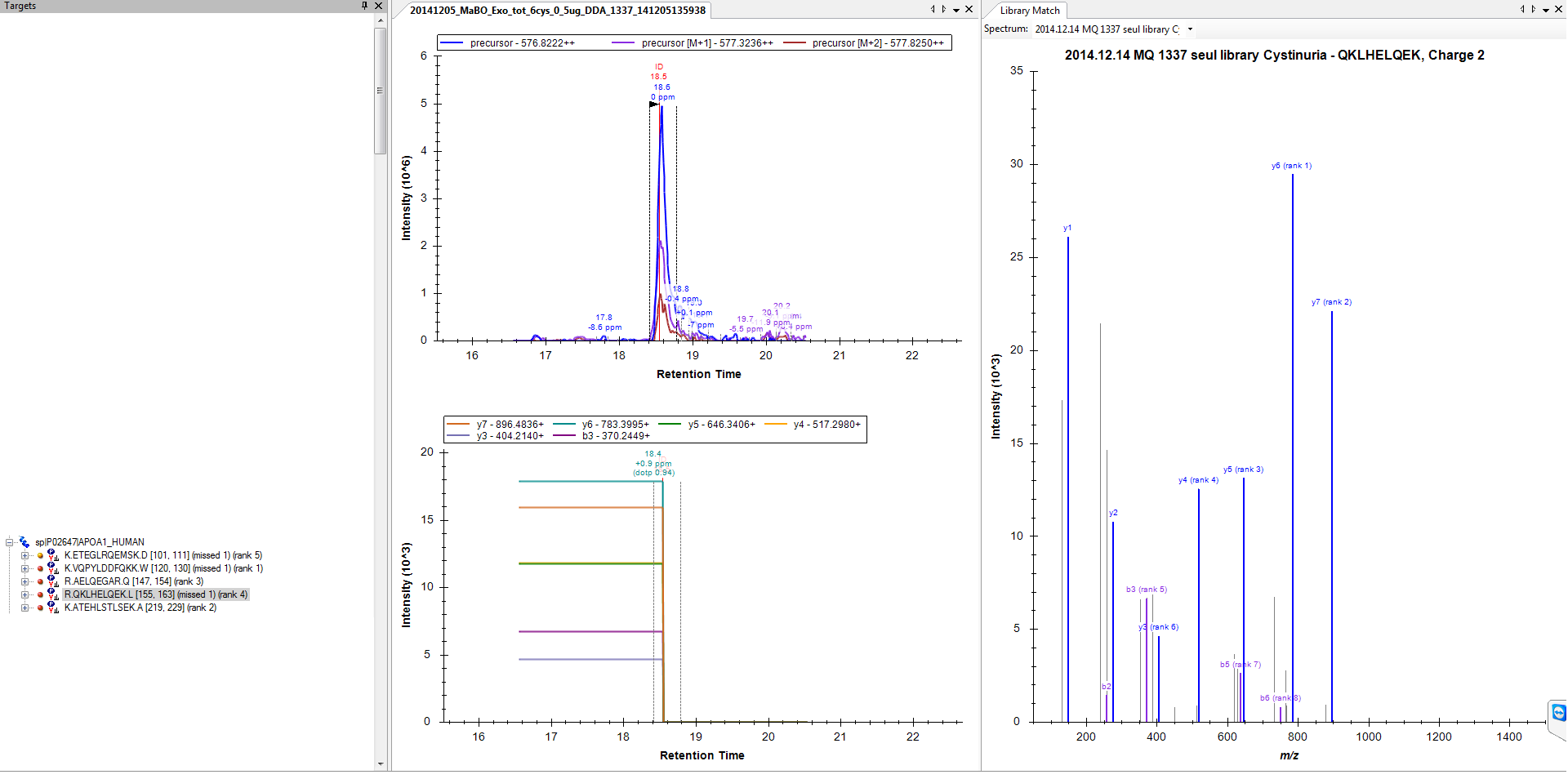 2014.12.14 Strange extraction.png
2014.12.14 Strange extraction.png 2015.02.03 Not smooth PRM.png
2015.02.03 Not smooth PRM.png