| jdemeter responded: |
2022-07-29 12:27 |
Alternatively, is there a way to use the annotations from the fasta file only - and not use the feature in Skyline that gets them from uniprot?
Thanks again,
Janos
|
| |
| Brian Pratt responded: |
2022-07-29 14:36 |
Hi Janos,
Skyline only goes to Uniprot if the FASTA header doesn't already have the annotations in it. So if you can get the gene name into the FASTA in the first place, none of this should be necessary. That should look something like this:
sp|P49768|PSN1_HUMAN Presenilin-1 OS=Homo sapiens OX=9606 GN=PSEN1 PE=1 SV=1
Thanks for using the Skyline support board,
Brian Pratt
|
| |
| jdemeter responded: |
2022-08-03 19:14 |
Hi Brian,
Thank you for getting back to me. I am using ncbi protein ids. I tried to format them like uniprot, e.g.:
NP_000007 acyl-CoA dehydrogenase medium chain GN=ACADM OS=Homo sapiens OX=9606
but it doesn't work - I am getting the uniprot annotations.
Thanks,
Janos
|
| |
| Brian Pratt responded: |
2022-08-04 10:48 |
If you can provide an example of your data, I'm sure we can figure something out. If it's small, you can attach it to your reply here, or you can upload to http://skyline.ms/files.url (or anything else you find convenient and/or suitably private).
Best regards,
Brian
|
| |
| jdemeter responded: |
2022-08-05 13:39 |
|
| |
| Brian Pratt responded: |
2022-08-08 14:29 |
Thanks for supplying the data.
For better or worse, when Skyline detects gaps in the metadata for a protein, it goes to Uniprot for details - and then prefers those details to the ones it started with in the FASTA file. You can make the case that this is backwards, I suppose - that only the gaps should be filled in, and the original file values should left alone, but this is the way we've always done it.
The header format which Skyline will interpret as complete, requiring no web access, looks like this:
SP|NP_000005|A2MG_HUMAN alpha-2-macroglobulin OS=Homo sapiens OX=9606 GN=A2M
That gives you a protein name "SP|NP_000005|A2MG_HUMAN" and puts the "NP_000005" as the accession and "AM2G_HUMAN" as the preferred named. You can then set your document to view proteins by accession, of course. Note the order OS, OX, GN, which is a defacto standard (and is what Skyline is expecting).
I hope this helps,
Brian Pratt
|
| |
| jdemeter responded: |
2022-08-09 10:13 |
Thank you very much, Brian!
Janos
|
| |
| jdemeter responded: |
2022-08-11 11:46 |
Hi Brian,
I tried what you suggested but sorry to say, it still doesn't work. For this protein:
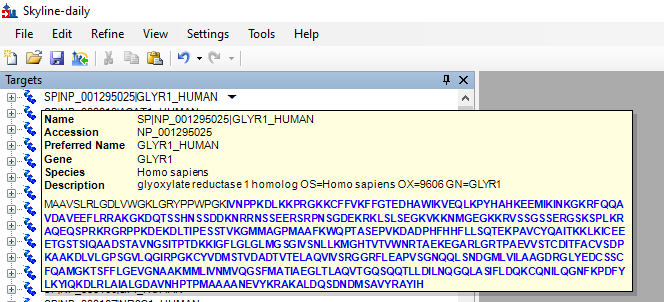
SP|NP_001295025|GLYR1_HUMAN glyoxylate reductase 1 homolog OS=Homo sapiens OX=9606 GN=GLYR1
it shows the expected name, accession and preferred name, but the gene is '<name: SP|NP_001295025|GLYR1_HUMAN>'.
For another example:
SP|NP_000010|ACAT1_HUMAN acetyl-CoA acetyltransferase 1 OS=Homo sapiens OX=9606 GN=ACAT1
the name is correct, but the accession is P24752, the preferred name is THIL_HUMAN and for gene it shows 'ACAT1 ACAT MAT'.
I attached the library I am using. (I have skyline-daily 22.1.9.208).
Thank you for your help,
Janos
|
|
| |
| Brian Pratt responded: |
2022-08-11 13:07 |
This is perplexing. When I load your file into Skyline-daily (same version) it seems to work as expected - see attached screen grab. Is there any chance at all that you're not using the fasta file that you mean to be using?
|
|
| |
| jdemeter responded: |
2022-08-11 13:21 |
I am importing the dataset via the 'import dda peptide search' option and select the fasta library in the last step of the import. I double-checked it - I am importing the same file I sent you.
Thanks,
Janos
|
| |
| Brian Pratt responded: |
2022-08-11 13:41 |
Can you verify that just importing the fasta directly (File>Import>FASTA menu, or just copy+paste into Targets window) works as expected?
If so, there's apparently something about the 'import dda peptide search' option that behaves differently. If that's the case, our quickest path to figuring this out would be if you could provide the files you're using at each step.
Thanks
Brian
|
| |
| jdemeter responded: |
2022-08-11 14:19 |
|
| |
| Brian Pratt responded: |
2022-08-12 08:14 |
Excellent information, that narrows things down considerably. We're looking into it.
Thanks,
Brian
|
| |
 fa.png
fa.png