| |
| Nick Shulman responded: |
2022-02-18 12:32 |
We have seen that error before. It happens if Skyline gets all the way to the end of the file that it is trying to extract chromatograms from, and Skyline has not used any of the spectra.
One of the reasons that this error might happen is if the "Retention Time Filtering" setting at "Settings > Transition Settings > Full Scan" says to use spectra within X minutes of the MS/MS IDs, but the retention times of the IDs in your spectral library are a completely different magnitude compared to the retention times that can actually be found in your .raw file.
You can find a little bit more information about that here:
https://skyline.ms/announcements/home/support/thread.view?rowId=54687
If you are still having trouble, you can send us your Skyline document and your raw file if you would like.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including spectral libraries.
You can also package up your .d folder in a zip file.
If those two zip files are less than 50MB you can attach them to this support request.
You can upload larger files here:
https://skyline.ms/files.url
-- Nick |
| |
| af1234 responded: |
2022-02-18 14:58 |
Hi Nick,
it worked by changing the triggered chromatographic acquisition time from empty to my gradient length. I take the chance to ask two follow up questions. For Mprophet, how do I select only the peptides having a qvalue of less than 1% and then export the light/heavy ratios? I added the decoys and generated an mprophet model but now I am a bit unsure on how to proceed. When I check the model qvalues there are a lot of 0s and the highest value is only e-4. |
| |
| Nick Shulman responded: |
2022-02-18 15:18 |
When you go to:
Refine > Reintegrate
there is a checkbox to tell it to only integrate peaks whose Q value is better than X%.
If you check that checkbox, then all of the low confidence results will have #N/A as their area instead of the actual area.
The other way to filter out high Q-values is to add the "Detection Q Value" column to your report.
If you want to learn more about the Document Grid and custom Reports in Skyline you can take a look at this tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_custom_reports
There are situations where you train an mProphet model, and all of your target peptides end up with really good q-values, even though they should not have such good q-values. What has happened in those situations is that the algorithm has managed to find a set of features that are really good at distinguishing decoy peptides from target peptides, but those features are not actually distinguishing true identifications from false identifications. One way that this can happen is if the spectral library was built from the same results that you are extracting chromatograms from. The extracted chromatograms match the target peptides in the library because it's the same data, and the decoy peptides do not match because the decoy peptides are not in the library.
One thing that I always recommend if you are training an mProphet model is to choose "Include all matching scans" for the Retention time filtering at "Settings > Transition Settings > Full Scan". If you extract short chromatograms, every candidate peak that Skyline looks at will by construction be really close to the predicted retention time, which causes the features to not get the weights that they should have.
If you send me your Skyline document I could probably give you more advice.
-- Nick |
| |
| af1234 responded: |
2022-02-19 10:14 |
Hi NIck,
It makes sense for the FDR calculation. Should I then import a FASTA just to get all the peptides and avoid using a library altogether? Our spectral library was indeed generated on the same data by direct DIA.
If you can have a look to the document would be great. Basically, we are seeing the heavy at the time 0 which is impossible as the animals were sacrificed immediately after feeding. |
| |
| Nick Shulman responded: |
2022-02-19 14:08 |
Did you intend to attach a file to this support request?
It might be that the file you tried to attach was larger than the 50MB size limit.
You can upload larger files here:
https://skyline.ms/files.url
--Nick |
| |
| af1234 responded: |
2022-02-21 15:02 |
Hi Nick,
I uploaded the skyline document. Let me know if you have any insights. |
| |
| Nick Shulman responded: |
2022-02-21 21:59 |
Thank you for sending your Skyline document.
I see that the peak scoring model that you have trained used the "Default" model. We usually recommend that you choose "mProphet" in the "Choose model" dropdown. When you choose "Default" for the model, the weights of the features never change relative to each other: all that Skyline does is figure out how to scale the weights so that the scores of the decoy peptides have a normal distribution centered at zero.
When you choose "mProphet" for the model, the weights end up getting chosen so as to maximize the separation between the decoys and targets.
If you look at the peak scoring model, the scores for the decoys and targets almost perfectly overlap with each other in a pair of gaussians centered around 7 (attached screenshot "PeakScoringModel.png"). This is a little confusing because Skyline is also showing you the "Decoy normal distribution" which is centered at zero. I am not sure exactly what it means when the actual distribution of decoy scores is nowhere near the "Decoy normal distribution". I think that might mean that the model was trained on a completely different set of data, and, for this reason, the weights and offset do not cause the scores in this dataset to be in the correct place.
I would recommend that you choose "mProphet" for the model, and then press the "Train Model" button. When I do that, I get something which looks pretty good ("mProphetTrainedModel.png"). The scores for most of the target peptides overlap with the decoy peptides, but there is a definite set of target peptides to the right which have significantly higher scores.
If you train an mProphet model and use it to reintegrate the peaks in your document, I think you will end up with DetectionQValue values that you can trust, at least in terms of how good the light version of the peptides look.
There are a bunch of scores such as "Reference intensity dot-product" or "Standard library dot-product" which are unavailable right now. These scores are shown in gray and italics, which indicates that some, but not all of the peptides in your document have no value for those features.
If you select one of the gray italicized scores in the "Available feature scores" grid, you can select the "Feature Scores" tab and see a bar graph of the distribution of the values for that particular feature. If you hover the mouse below the "unknown" bar on the graph, a binoculars button will appear and you can click on that button. When you do that, the peptides in your document which are missing that score will be listed in the "Find Results" window in Skyline.
You can use the Find Results window to select and then delete the peptides which are missing these scores. It seems you have some peptides in your document which only have the light form of the peptide and no heavy form. For this reason, when you train a model, Skyline does not look at the features related to heavy peptides, because a trained model can only use features which are present for all peptides in the document.
-- Nick |
|
| |
| af1234 responded: |
2022-02-22 18:46 |
Hi Nick,
thanks a lot for the explanation. Now to try increase the number of peptides we can see, I am trying with a fasta file with pretty strict parameters (only unmod and prototypic without missing cleavages) but when I try to import a file after generating the decoys I get this error (which I have seen on the forum but without a solution). If I import it without decoys it works.
Skyline version: 21.2.0.369-2efacf038 (64-bit)
Installation ID: 93868471-5719-4cec-a968-fe7567dcbbbb
Exception type: ArgumentException
Error message: An item with the same key has already been added.
--------------------
System.ArgumentException: An item with the same key has already been added.
at System.ThrowHelper.ThrowArgumentException(ExceptionResource resource)
at System.Collections.Generic.Dictionary`2.Insert(TKey key, TValue value, Boolean add)
at pwiz.Skyline.Model.SrmDocument.OnChangingChildren(DocNodeParent clone, Int32 indexReplaced) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Model\SrmDocument.cs:line 798
at pwiz.Skyline.Model.DocNodeParent.ChangeChildren(IList`1 children, IList`1 counts, Int32 indexReplaced) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Model\DocNode.cs:line 1243
at pwiz.Skyline.Model.SrmDocument.ChangeSettingsInternal(SrmSettings settingsNew, SrmSettingsChangeMonitor progressMonitor) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Model\SrmDocument.cs:line 1113
at pwiz.Skyline.SkylineWindow.ImportResults(SrmDocument doc, String nameResult, IEnumerable`1 dataSources, OptimizableRegression optimizationFunction) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\SkylineFiles.cs:line 2920
at pwiz.Skyline.SkylineWindow.ImportResults(SrmDocument doc, List`1 namedResults, String optimize) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\SkylineFiles.cs:line 2869
at pwiz.Skyline.SkylineWindow.ModifyDocumentInner(Func`2 act, Action onModifying, Action onModified, String description, Func`2 logFunc, AuditLogEntry& resultEntry) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Skyline.cs:line 792
at pwiz.Skyline.SkylineWindow.ModifyDocumentOrThrow(String description, IUndoState undoState, Func`2 act, Action onModifying, Action onModified, Func`2 logFunc) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Skyline.cs:line 761
at pwiz.Skyline.SkylineWindow.ModifyDocument(String description, IUndoState undoState, Func`2 act, Action onModifying, Action onModified, Func`2 logFunc) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Skyline.cs:line 738
at pwiz.Skyline.SkylineWindow.ImportResults() in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\SkylineFiles.cs:line 2740
at System.Windows.Forms.ToolStripItem.RaiseEvent(Object key, EventArgs e)
at System.Windows.Forms.ToolStripMenuItem.OnClick(EventArgs e)
at System.Windows.Forms.ToolStripItem.HandleClick(EventArgs e)
at System.Windows.Forms.ToolStripItem.HandleMouseUp(MouseEventArgs e)
at System.Windows.Forms.ToolStrip.OnMouseUp(MouseEventArgs mea)
at System.Windows.Forms.ToolStripDropDown.OnMouseUp(MouseEventArgs mea)
at System.Windows.Forms.Control.WmMouseUp(Message& m, MouseButtons button, Int32 clicks)
at System.Windows.Forms.Control.WndProc(Message& m)
at System.Windows.Forms.ToolStrip.WndProc(Message& m)
at System.Windows.Forms.ToolStripDropDown.WndProc(Message& m)
at System.Windows.Forms.NativeWindow.Callback(IntPtr hWnd, Int32 msg, IntPtr wparam, IntPtr lparam)
Exception caught at:
at System.Windows.Forms.Application.ThreadContext.OnThreadException(Exception t)
at System.Windows.Forms.Control.WndProcException(Exception e)
at System.Windows.Forms.NativeWindow.Callback(IntPtr hWnd, Int32 msg, IntPtr wparam, IntPtr lparam)
at System.Windows.Forms.UnsafeNativeMethods.DispatchMessageW(MSG& msg)
at System.Windows.Forms.UnsafeNativeMethods.DispatchMessageW(MSG& msg)
at System.Windows.Forms.Application.ComponentManager.System.Windows.Forms.UnsafeNativeMethods.IMsoComponentManager.FPushMessageLoop(IntPtr dwComponentID, Int32 reason, Int32 pvLoopData)
at System.Windows.Forms.Application.ThreadContext.RunMessageLoopInner(Int32 reason, ApplicationContext context)
at System.Windows.Forms.Application.ThreadContext.RunMessageLoop(Int32 reason, ApplicationContext context)
at pwiz.Skyline.Program.Main(String[] args) in C:\proj\skyline_21_2_x64\pwiz_tools\Skyline\Program.cs:line 308 |
| |
| Nick Shulman responded: |
2022-02-22 19:03 |
That's an interesting error. We have seen that error reported many times over the past few years, but, so far, no one has included their email address with the error report so I have never been able to follow up and find out what causes it.
It appears the error happened when you did "File > Import > Results", but I am thinking that the real problem might have happened just before that. What was the thing that you did right before you did "File > Import > Results"? The error seems to be saying that the same peptide appears in two different locations in the Targets tree. Skyline was supposed to make a copy of an object, but instead has put the exact same object in two places.
It would be helpful if you could send me your Skyline document again ("File > Share" to create a .zip file).
It sounds like you might have also done something with a FASTA file right before that, or maybe changed some peptide filtering settings, or something. Please send me your FASTA file and let me know what else you remember doing right before this error happened.
By the way, if you were to exit Skyline and open up the same document again, I imagine the error will stop happening. This error just involves the in-memory representation that Skyline has for your document, and if Skyline were to load the document from disk again, everything would be as it is supposed to be. But if you send me your document and your FASTA file, I'm thinking I might be able to figure out how to make it happen again.
-- Nick |
| |
| af1234 responded: |
2022-02-23 07:33 |
Hi Nick,
That makes a lot of sense.
From the N15 file, I remove all peptides and results and then changed peptides settings and imported the fasta (same one used as background proteome, so maybe that is the issue?). Then added the decoys and imported the results. I will upload the document in its current state as the error is reproducible by importing any pasef file. |
| |
|
|
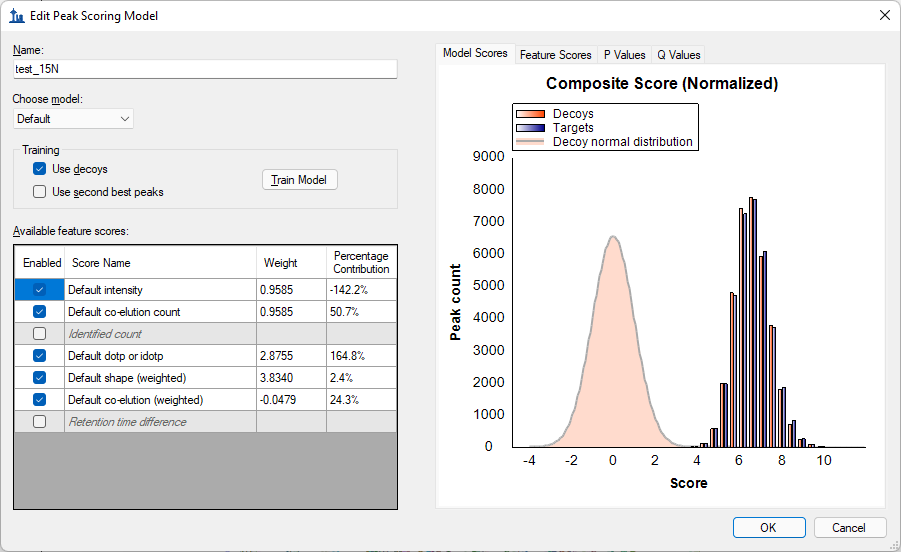 PeakScoringModel.png
PeakScoringModel.png mProphetTrainedModel.png
mProphetTrainedModel.png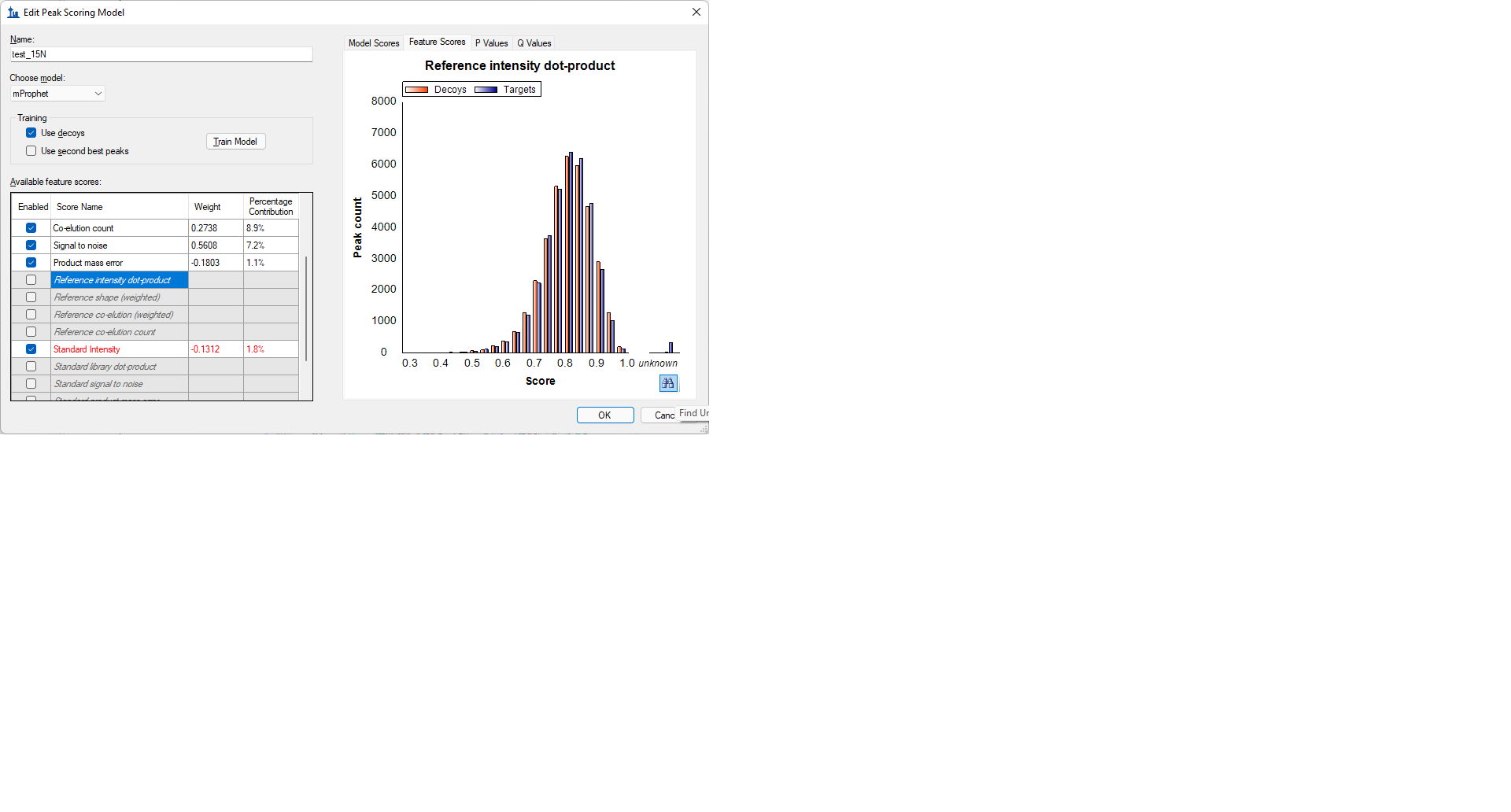 FindUnknownPeptides.png
FindUnknownPeptides.png