Hi Skyline Team,
Please see attached a document which is giving some odd behavior when using iRT in small molecule mode. I built a retention time predictor using 11 analytes (stable isotope labeled spike-ins) in the sample, then used a few runs to build an iRT library for about 50 other compounds. This seems to have gone really well. I then imported additional data and told it to import only 1 minute around the predicted RT, which it did. However, there are still a lot of cases where Skyline integrates ('picks') the incorrect peak, even though there is a very nice peak directly at the 'predicted retention time'. This is easily visualized because you have the nice option to show the Predicted Retention Time in the chromatogram window.
Document attached. Hoping there's just a button i neglected to check somewhere that tells Skyline to actually use the predicted retention time that it has access to, to help pick the right peak. Otherwise, i just can't understand why Skyline wouldn't be using the information available. Right now it seems that even though the predictor is enabled, it's pretty much doing the same thing it would if you gave it no RT information at all.
Thanks!
Will
|
| |
| Nick Shulman responded: |
2021-09-29 13:31 |
Will,
Can you tell us an example of a molecule and replicate that we should look at in that Skyline document?
-- Nick |
| |
| Will Thompson responded: |
2021-09-30 07:14 |
There are a bunch of them, but for a couple examples (make sure right click/retention time prediction is turned on in the chromatogram window)
1. Click on Asparagine. Click on replicate ID64_01 and _02 as well as ID69_01 and _02. Both cases there is a decent peak at the predicted RT that is ignored. (although not the largest in the chromatogram)
2. Dimethylglycine is egregious. Half the replicates Skyline picked the incorrect peak, even though there is a beautiful peak spot on the predicted RT in every case.
3. Glutamine makes the mistake in one case.
4. Isoleucine/Leucine/Norleucine is a really important test case. There are three closely eluting, isobaric compounds which are a difficult 'automation' integration case. Same molecular weight, but very distinct peaks and the iRT prediction is doing a great job of tracking them. Click on Isoleucine (which should be the first peak of the three), and click on replicate P12_ID64_01. You'll see that the predicted RT is dead in the center of the correct peak, but Skyline picks the largest peak (the second one) which is leucine. This leads to the requirement to manually correct 100% of the data. Norleucine is the third (very small) peak. The iRT also predicts its retention time correctly, but still Leucine (the middle peak) is selected by Skyline.
Basically, it seems like Skyline is just choosing the most abundant peak with reasonable idotp and ignoring the iRT prediction. I think it
Also i just realized that in the document that i uploaded, replicate "P12_ID79_02" is a little squirrelly. Just edit/manage results remove that replicate.
I have a talk at ASMS which is using iRT prediction for metabolomics as one method of improving 'FDR' for metabolite ID, so it would be awesome if we can get this looked at soonish...otherwise i can 'fix' the integration manually since skyline is ignoring the predicted retention time...but that feels a little like cheating. The unique angle here is that this is Capillary Electrophoresis data and LC (these are really migration times not retention times).
Cheers
W |
| |
| Will Thompson responded: |
2021-09-30 07:17 |
I should also state that the replicates P12_ID84_01 through _04 were what was used to train the iRT database. Those were already manually curated in the document before the other data (which would require prediction) was imported. So that is why the ID84 replicates are always at the correct RT. |
| |
| Nick Shulman responded: |
2021-09-30 09:32 |
Will,
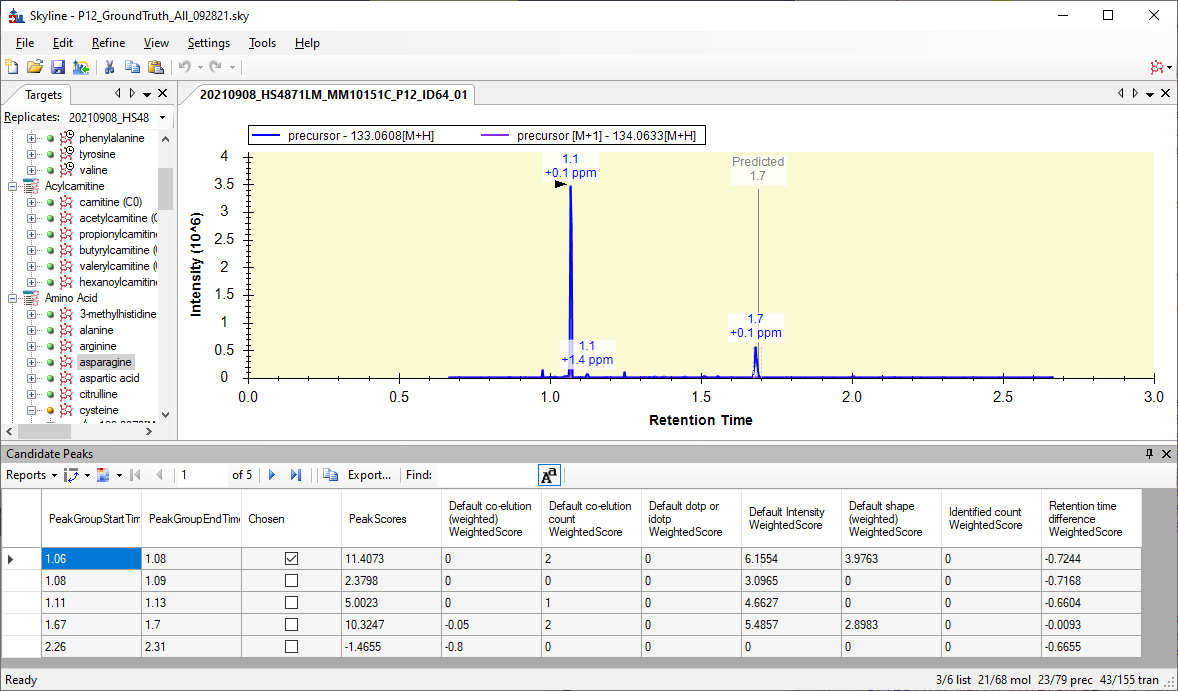
I am working on a new feature for Skyline: the Candidate Peaks grid which will show you the peaks that Skyline considered before choosing the best peak.
You can see in the attached image that the peak that Skyline chose got a score of 11.4, and the peak that you would have liked Skyline to have chosen got a score of 10.3.
The peak that Skyline chose did much better on "Intensity" and "Shape", and did worse on "Retention time difference".
If only Retention time difference were weighted much higher than it actually was, Skyline would have chosen the peak you wanted.
I wonder if things would be improved if Skyline gave you the ability to tweak the weights in the default peak scoring model. I am attaching a .sky.zip of a new document which has a model where the retention time difference score is weighted more. In order to set those weights I had to hand-edit the .sky file in a text editor.
I think our default set of weights were chosen based on looking at datasets with much longer gradients. In those cases, a one minute difference from the predicted retention time should not penalize the score too much, but with your short gradient data, the weights that we have chosen are not helpful.
I will ask around and see what we might be able to do about making this better this version.
-- Nick |
|
| |
| Will Thompson responded: |
2021-09-30 12:12 |
Hi Nick
Thank you for giving me a bit more of a peek under the hood with this one. I think the basic answer to your question is yes, it definitely needs to weight the predicted retention time more heavily, and I think the model could be made more robust if it utilized the averaged measured peak width within the data instead of a 'fixed' window of some timeframe. Separations (even by LC) are getting a lot faster and i think this could explain why Skyline generally doesn't do as well with really nice (fast) chromatographic separations as one would expect. If it is not penalizing something which is 20 peak widths away because it is still within 1 minute, it seems like the algorithm just isn't making the most of the data available to it for the purposes of scoring. Something like downweighting peaks from x1 to x0 as they go to >2-4 average peakwidths away from the predicted RT might make sense...
Maybe instead of asking (and requiring?) the user to 'tweak' the scoring weighting, Skyline could enable the user to define an expected peakwidth (width at base, wb) and use this to somehow weight the factors appropriately on the back-end?
It looks like your weighting modifications were able to fix the errors for what i would call "egregious" errors, where the peak was clearly way wrong in RT and just lower abundance than the most major peak. This is a big step forward, thank you! It looks like the changes did not make any difference for the Iso/Leu/Norleucine peak selection...still not recognizing isoleu is first and just before leucine (which has an iRT standard). Maybe it would be worth taking a look at the weightings to see if there are any efforts that can be made in the current model which can handle this case reliably? This seems like a potentially difficult situation which may cause a rethink of the algorithm. I will note that this would be the same case for phosphopeptides which a different site is modified on the peptide...small shifts in RT with the exact same MW. So this scenario is not unique to small molecules and could be important in many cases.
Thanks for all the work on this! This first round was such a large improvement, how difficult would it be to apply the new weightings for an automated analysis of a much larger dataset (approx 5 x the number of runs). Or, if i import that data into the skyline file you sent me, will it apply the 'new' weightings to the new runs? :)
Cheers
Will |
| |
|
|
 GroundTruthPeakScores.png
GroundTruthPeakScores.png