| Nick Shulman responded: |
2020-10-05 08:35 |
The problem is that you have told Skyline only to extract MS2 chromatograms from DDA data.
In a DDA experiment, the mass spectrometer does not have a reliable schedule for selecting precursors for fragmentation, so the MS2 chromatograms look the way you are seeing them, with long straight lines connecting the points where the precursor happens to have been selected.
What you should do is tell Skyline that you want MS1 chromatograms.
The way to do that is:
1. Go to "Settings > Transition Settings > Filter" and add "p" to the list on "Ion types". (So, maybe, make it so the text in the Ion Types box is "p, y, b")
2. Go to "Settings > Transition Settings > Full Scan" and choose something other than "None" for "Isotope peaks included".
3. Also on the Full Scan tab, change "Acquisition method" to "DDA". (you currently have it set to "Targeted").
After you have done this, you need to reimport your chromatograms by going to:
Edit > Manage Results > Reimport
After you have done that, you will have nice looking MS1 chromatograms. Also, because you have set the MS/MS filtering acquisition method to "DDA", the MS2 chromatograms that you are currently seeing will be displayed in dotted lines indicating that they should not be used for peak integration.
It might be helpful to take a look at the MS1 full scan filtering tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_ms1_filtering
-- Nick |
| |
| yingzhu1992 responded: |
2020-10-05 09:47 |
Dear Nick,
Thanks for your reply.
my raw data was measured by targeted aqusition method.
I tried the settings that you showed me. if I changed the acquisition method to "DDA", Skyline showed "Chromatogram information unavailable".
If I only changed the Ion type, the peaks are still incomplete. do you have any ideas about this?
Thanks for your help!
Ying |
| |
| Nick Shulman responded: |
2020-10-05 10:06 |
Can you send us that raw file you were looking at ("L1_20200930_YZ_31_7.raw")?
Files that are less than 50MB can be attached to this support request.
You can upload larger files here:
https://skyline.ms/files.url
Did you reimport ("Edit > Manage Results > Reimport") after you made the changes to your Transition Settings? Anytime you change the set of transitions in your Skyline document you need to reimport in order to tell Skyline to extract the chromatograms again. Otherwise, these new chromatograms will say "Chromatogram information unavailable".
Was your targeted acquisition method a scheduled method? That is, did you tell the mass spectrometer to collect data for a particular precursor over a specific amount of time?
In your attached picture, "Raw data.PNG", it seemed to me that you were looking at an MS1 chromatogram. (I am not sure, because I am not very familiar with Thermo's QualBrowser). If you had a scheduled targeted method, then I would expect your MS2 chromatograms to be truncated to only the times over which you told the mass spectrometer to collect data. Also, if the MS/MS Acquisition Method is "DIA" or "Targeted" (i.e. anything other than "DDA"), then Skyline will truncate the MS1 chromatograms so that they match the MS2 chromatograms.
-- Nick |
| |
| Nick Shulman responded: |
2020-10-05 13:00 |
Ying,
I see that you already did upload the file "L1_20200930_YZ_31_7.raw".
That file really does look like a Data Dependent Acquisition file. Unique precursor m/z values only get isolated a single time, based on what the mass spectrometer sees in a particular MS1 scan. So, the chromatogram in your screenshot consists of the following scans:
| scan number |
retention time |
precursor m/z |
| 8749 |
36.97858258 |
576.30621 |
| 8760 |
37.01039304 |
576.30701 |
| 8769 |
37.03713451 |
576.30664 |
| 8777 |
37.06086131 |
576.30634 |
| 22616 |
77.84435621 |
576.30463 |
-- Nick
|
| |
| yingzhu1992 responded: |
2020-10-05 13:53 |
Dear Nick,
Thanks for your reply.
I did reimport after I changed transition settings. If I only changed the ion types, it looked same as previous; when I changed the ion types and acquisition method to DDA , then Skyline said "Chromatogram information unavailable". What should I do to solve this and tell Skyline that I want MS1 chromatograms.
My data used the method named SureQaunt. The MS1 is a full scan, but the MS2 is triggered based on the targeted mass list without scheduled retention time. when the heavy labeled peptides were detected, then the light peptides were triggered to be measured.
Yes, the attached "Raw data.PNG", it is an MS1 chromatogram.
Thanks for you help.
Ying
|
| |
| Nick Shulman responded: |
2020-10-05 14:09 |
For SureQuant methods, we recommend that you use Skyline-Daily instead of Skyline 20.1.
You can install Skyline-Daily from here:
https://skyline.ms/project/home/software/Skyline/daily/begin.view
We have added a new feature in Skyline-Daily specifically to make analyzing SureQuant data better. This feature is a "Triggered Acquisition" checkbox on "Settings > Transition Settings > Instrument", which change the way that Skyline does background subtraction and peak picking so that it does not get tripped up by the fact that the MS2 chromatograms are truncated. You can learn more about this feature here:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=TriggeredAcquisition
We are going to release Skyline 20.2 in a week or so, and that will include this SureQuant support. For now, you should use Skyline-Daily.
I am not sure I understand the other problems that you are running into. If you are still having problems using Skyline-Daily, can you send me a new .sky.zip with what you are currently looking at?
-- Nick |
| |
| yingzhu1992 responded: |
2020-10-06 09:48 |
Hi Nick,
I tried to use the Skyline-Daily. And I also checked some Q&A about SureQuant method in Skyline website. I also changed "full scan" -> DIA acquisition method and clicked on the Triggered acquisition. But an error happened when importing the raw data.
Thanks for your help.
Ying |
|
| |
| Nick Shulman responded: |
2020-10-06 10:41 |
Thank you for reporting this error. I will make sure that it is fixed in the next update of Skyline-Daily.
-- Nick |
| |
| sponce1 responded: |
2020-10-15 10:54 |
Hi Skyline, yeah i'm having a similar problem. I performed a PRM standard curve on a Exploris 480 for 6 heavy peptides. Skyline wont extract the full chromatogram see my screenshot in the attached word doc, the left screen is Skyline and the right is Skyline daily.
I also uploaded the following files "20201002.zip" which are my raw files, "HEAVY6_PRM.sky" my skyline document and "PRM.sky" which I made using Skyline Daily.
Thank you!!!
-Sean |
|
| |
| Nick Shulman responded: |
2020-10-15 11:08 |
Sean,
Can you upload .sky.zip files instead of the .sky files that you did upload?
In Skyline you can use the menu item:
File > Share
to create a .sky.zip file containing your Skyline document and supporting files including extracted chromatograms and spectral libraries.
Thanks,
-- Nick |
| |
| sponce1 responded: |
2020-10-15 13:14 |
Done.
Thanks Nick!
-Sean |
| |
| Nick Shulman responded: |
2020-10-15 21:35 |
Sean,
I am not sure what you would like Skyline to do differently.
The chromatograms that Skyline gives you have points from all of the spectra whose isolation window matches the precursor in your Skyline document.
If your mass spectrometry method only starts collecting MS2 scans after the precursor has been seen in MS1, the MS2 chromatograms will inevitably be truncated.
In Skyline 20.2 we added the feature "triggered acquisition" which will alleviate some of the negative aspects of this truncation: background subtraction, and peak boundaries that extend across times where no data was acquired.
You can learn more about this feature here:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=TriggeredAcquisition
Let me know if I'm misunderstanding something.
-- Nick |
| |
| sponce1 responded: |
2020-10-19 18:21 |
do not remove/cut data. i always thought skyline was a nice data extraction program not a data manipulator. if as mentioned above you extract a ms2 scan in qual browser on thermo instrument produced data, you can see a nice peak. but skylines new update show massive data elimination so much so that peak integration is meaningless (see attached screenshots), this happened with the data I uploaded above.
the other thing that has me wondering is the new 480 has a super fast scan speed, 24 hz. so i wonder if your old algorithms cant handle data smoothing at that fast a rate? like basically just extract it and let the data speak towards continuity or smoothness
thanks !
-Sean |
|
| |
| Nick Shulman responded: |
2020-10-19 19:32 |
Sean,
I do not see any chromatograms that look like your latest screenshots. Which peptide is that? Can that be found in the .sky.zip's that you sent earlier? If not, could you send me another .sky.zip?
What does "TAS" mean? Does that mean having the "Triggered Acquisition" checkbox checked?
When "Triggered Acquisition" is selected, Skyline tries to guess how long of a gap between scans corresponds to a time when the mass spectrometer is not collecting data for your peptide of interest. In order to decide on a gap interval, Skyline looks at the median time between scans and multiplies that median time by ten, and that is the gap interval. If any scans are more than that interval apart, then Skyline breaks the chromatogram in between those scans.
The choice of using the median and the number 10 were fairly arbitrary. If there was a lot of variability in terms of how quickly the mass spectrometer could acquire scans for a particular analyte, then I could imagine that Skyline might be tricked into thinking the mass spectrometer was not collecting data for a particular analyte at a particular time, even though the mass spectrometer was actively acquiring those scans as fast as it could.
I might need to tweak those numbers after I see your data.
-- Nick |
| |
| sponce1 responded: |
2020-10-20 12:31 |
Hi Nick,
Thanks so much for your response, and lets hope we can get this sorted.....
Yes TAS means with the "Triggered Acquisition" checkbox checked. I uploaded "20201002 Skyline PRM" to you. In it, there are two skyline documents and 2 skyline chromatogram data (if you also need the raw files I had previously uploaded that). The only difference is in one doc I have TAS off "HEAVY6_PRM", and the other doc TAS is ON "HEAVY6_PRM_TAS".
Many thanks!
-Sean |
| |
| Nick Shulman responded: |
2020-10-20 13:22 |
Sean,
I was not able to find a peptide which exactly matched what was in your PowerPoint, but the peptide GPSTPLPEDPNWDVTEFHTTPK in Replicate "4" looks similar.
There were a couple of places along the chromatogram where the mass spectrometer was able to collected one scan every 0.0035 minutes (i.e. 0.2 seconds).
The median time between scans in the chromatogram was 0.03114 minutes (i.e. 1.87 seconds).
Because of this, when Skyline has "Triggered Acquisition" turned on, Skyline thinks that any place where the time between successive scans matching that peptide is more than 18.7 seconds (i.e. 0.3114 minutes) is a gap where data was not being acquired for that peptide.
If it looks like Skyline is cutting out the parts of the chromatogram where the peptide is actually eluting, then I think that means that your SureQuant method is not triggering on the correct signal, because it seems to not be acquiring data at the exact times when it should.
I could imagine a scenario where the acquisition rate might be so variable that Skyline would need to allow you to customize some of the numbers (median; factor of 10) in the heuristic for deciding what is a gap.
Hope this helps,
-- Nick |
|
| |
| sponce1 responded: |
2020-10-20 18:45 |
yeah like make it more user defined available options fit for purpose as opposed to black box.
appreciated,
-sean |
| |
 Capture.PNG
Capture.PNG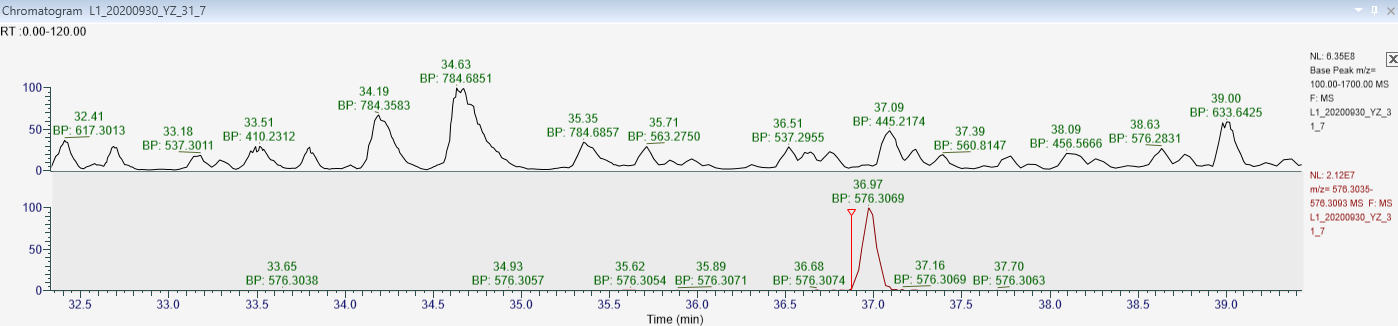 Raw data.PNG
Raw data.PNG Error.PNG
Error.PNG NoTriggeredAcquisition.png
NoTriggeredAcquisition.png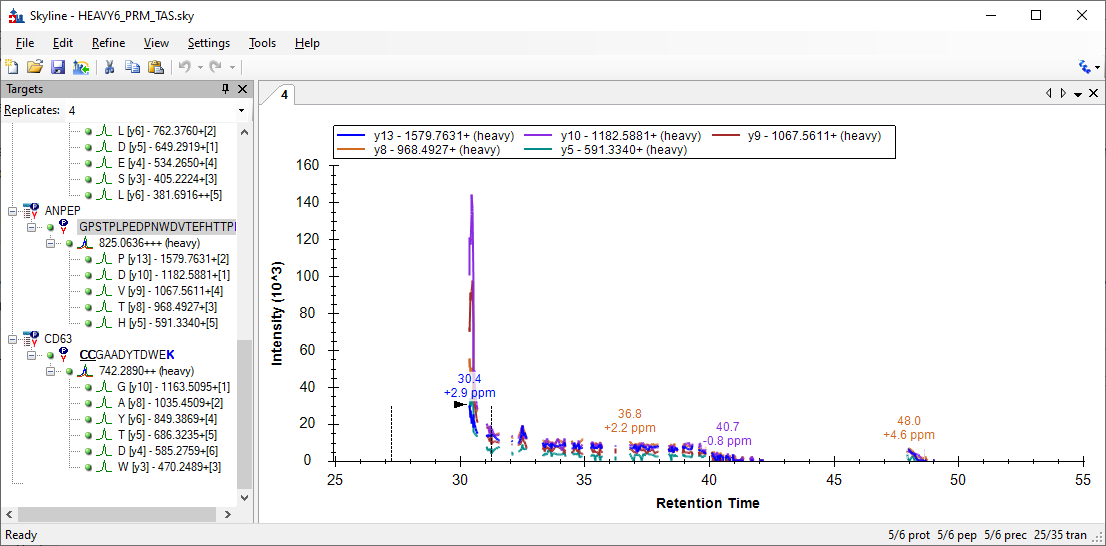 WithTriggeredAcquisition.png
WithTriggeredAcquisition.png