| |
| Nick Shulman responded: |
2020-10-01 09:21 |
Chinmaya,
The usual way to diagnose a problem like this would be to go to:
Refine > Reintegrate > Edit Current
and see if anything looks suspicious.
One thing that can happen when you add more replicates is that certain feature scores become unavailable to the model, because one or more peptides might not have a value for that feature in one or more replicates. This sometimes happens if some of your replicates were collected or imported with different mass spectrometry settings.
By the way, in Skyline-Daily, we added some new features related looking at the number of detections in your Skyline document. You can see these new graphs using the menu item:
View > Detections > Replicate Comparison
View > Detections > Histogram
I hope these new features work well for you.
You can always send us your Skyline documents and we can see whether there is anything suspicious.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If those .zip files are less than 50MB you can attach them to this support request.
Otherwise, you can upload them here:
https://skyline.ms/files.url
-- Nick |
| |
| Chinmaya k responded: |
2020-10-02 01:46 |
Hi Nick,
I was able to get rid of huge reduction in the number of peptides from Multi RAW file search by selecting Auto-calculate regression with (Time window - 5 min) a trained study specific iRT model generated using single RAW file search. However, the total number of peptides identified are quite low when compared with the search results from single RAW file seach.
I assume the reason for this is inaccurate detection of peptide RT with nearby transitions rather than the true transition peaks. For your reference, I have attached a screen shot of peptide (K.GILAADESVGTMGNR.L) peak identified in three technical replicates, where peptide RT is not accurately detected in two of the replicates though it is falling in the predicted RT range. There are several such peptides, where the peptide transitions were inaccurately detected at nearby RT in either of the replicates or files and therefore, the peptides is failing to pass the mProphet validation.
Are therer any methods to rectify the same?
I have also performed spectral library search for all the 18 rawraw files from 6 coditions. I have attached the screenshot of peptides (q-value < 0.01) identified in each file. This is resulting with a ~3500 non-redundant peptides (Cumulative).
For your reference, I have searched a single and mulitple (Three replicates of one condition) raw file seperately. The skyline documents for the same were uploaded in ( https://skyline.ms/files.url) under the description 1) DIA_Search_Single_RAW_File and 2) DIA_Search_One_Condition_With_Technical_Replicates
Let me know, if you need more information.
--
Chinmaya |
|
| |
| Nick Shulman responded: |
2020-10-02 08:20 |
Chinmaya,
The Time Window on the retention time predictor does not matter for choosing peaks. That setting only matters if you are exporting a scheduled method-- that's the time range around the predicted retention time where the mass spectrometer should be collecting data for a particular peptide.
I believe the problem is that in your single replicate document, the high number of positive IDs that you are seeing are not valid because you calibrated your iRT predictor using the data in that replicate. That is, if you look at the Retention Time Score to Run regression, it is a perfectly straight line with no outliers.
If your iRT predictor was calibrated from the same replicate that you are trying to train a model on, then there will always be a target peak whose "Retention Time Difference" is zero, but there will not necessarily exist a decoy peak that is that good. For this reason, the trained model will think that Retention Time Difference is a great way to distinguish between true peaks and decoys, but that will not be a valid thing to be doing.
I believe the correct thing to do is uncheck the checkbox next to Retention Time Difference in the Edit Peak Scoring Model dialog, and train the model. This will result in the number of positive matches going down, but that will be the more accurate result.
I am not sure whether there is another technique for making sure that your iRT prediction values are not biased by the data that you are using to train the model.
-- Nick |
|
| |
| Chinmaya k responded: |
2020-10-05 06:19 |
Nick,
Thank you so much for that information. It was quite helpful. Although, I was confused initially.
Since, I have acquired some single injection runs of Gas Phase Fractionated (GPF) DIA runs, I used peptides identified from direct proteome database search of GPF for the iRT model. Which is resulting with run to score RT regression with outliers as you have mentioned. The screenshot of the Score to Run regression is attached below.
Using the same workflow, I carried out the a spectral library search for 18 raw files. However, the reintegration using mProphet model is failing to perfrom peak scoring. The screenshot of the error and the error info in .txt file are attached for your reference. What might be the reason for this?
--
Chinmaya |
|
| |
| Nick Shulman responded: |
2020-10-05 08:18 |
Another user once ran into the error "Failed attempting to reintegrate peaks. The index X must be between Y and Z" a few years ago:
https://skyline.ms/announcements/home/support/thread.view?rowId=27885
In that user's case, the same peptide appeared in the document more than once, and those two peptides had a different set of precursors under it.
In the document that you sent me, the decoy peptide "GLPEGDVAIDFFVTPNVR" appears twice in your document, and one of those has just a charge +2 precursor under it, and the other has +2 and +3.
I am not sure how your document got that way. I think you could fix this problem by generating decoys again with the menu item:
Refine > Add Decoys
-- Nick |
| |
| Chinmaya k responded: |
2020-10-06 22:26 |
It seems like, I was clicking "Okay" to a heavy label modifications (Which is not defined in Skyline - Please find the attached screenshot) while importing a spectral library, which resulting with peptides with light and heavy label in the document. However, when I delete heavy labelled peptides from the document and continue with the reintegration, it is failing to perform. This issue was solved by clicking "Cancel" option for modifications while importing the spectral library to document.
Finally, the DIA spectral library search and reintegration seems to be working well. In order to gain more accurate identifications without losing many targtes, I have carried out two step mProphet based reintegration. Below are the steps I have followed.
a) First one was soon after importing the raw files, mProphet reintegration was performed without "Retention Time Difference" as you have suggested in your previous response. This step resulted with 40,993 peaks with Q-value < 0.01.
b) Later, the resulting peptides were calibrated against the iRT model. This resulted with score to run regression of 0.9673.
c) Finally, RT calibrated peptides were reintegrated again with "Retention Time Difference" and this resulted with 78,272 peaks with Q-value < 0.01. This step further improved the score to run regression to 0.9886
Let me know, how valid this approach is?
The screenshots of each step is available in the attached PowerPoint presentation. The Audit Log from Skyline is also attached below for more information.
--
Chinmaya |
|
| |
| Nick Shulman responded: |
2020-10-07 09:15 |
That does not sound like a valid approach.
The problem is that you cannot use the data from this experiment to get predicted retention times, and then use those predictions to improve your peak picking in that same experiment.
You need to find a source of predicted retention times that is not this experiment itself.
One thing that you could do is create a spectral library using Prosit.
Prosit is a web service which predicts retention times and fragmentation for peptides using machine learning. One big limitation is that it does not know about very many PTMS (I believe all it can handle is oxidized methionine and carbamidomethyl Cysteine)
In order to enable Prosit in Skyline, you have to first go to:
Tools > Options > Prosit
and choose something for "Intensity Model".
Then you can create a spectral library by going to:
Settings > Peptide Settings > Library > Build
and choose "Prosit" for the Data Source and choose the right thing for "iRT Standard Peptides" (which is "Pierce" for your experiment).
I am not sure how accurate Prosit retention time predictions are, but any inaccuracies will not be biased in terms of targets or decoys, so you should be able to trust the FDR that comes out of your trained model.
-- Nick |
| |
| Chinmaya k responded: |
2020-10-10 03:28 |
The DIA search against the Prosit predicted library is crashing in my system because of limited amount of RAM and Processor speed.
However, I have reanalyzed the data against the study specific DDA based spectral library with a chromatogram library generated using Gas Phase Fractionated data.
Below is the workflow I have applied in this reanalysis;
1. Generating Chromatogram Library: 6GPF data (4m/z window)> PeCan search against Human Proteome database in Walnut(EncyclopeDIA) > Result > Chromatogram Library > Import to Skyline > Set the parameters under Peptide and Transition settings > Add Decoy > Match with DIA (25m/z window) raw data > Add resulting peptides to iRT model > Calibrate against standard iRT peptides > Save the model
2. Generating Empirically generated chromatogram library: Convert Prosit predicted peptide library for human to EncyclopeDIA compatible library using iRT model from previous step > Search all 6 GPF data against the empirically corrected library from Prosit > Result > Chromatogram Library > Import to Skyline > Set the parameters under Peptide and Transition settings > Add Decoy > Match with DIA (25m/z window) raw data > Add resulting peptides to new iRT model > Calibrate against standard iRT peptides > Save the new model
3. New Skyline Document > Set the parameters under Peptide and Transition settings > Import DDA based spectral library > Add the iRT model from step 2 > Add Decoy > Import all the DIA raw data > Calibrate the peptide RTs using iRT model > Keep peptide with minimum 6 transitions > Reintegrate using mProphet model > Export peptide transitions with 1% FRD (Q-Value < 0.01)
This approach is resulting with varying number of peptides and proteins with 1% FDR for different number of input RAW data. The search of single RAW file is resulting with 11,000 peptides corresonding to 2,200 proteins, while other two technical replicates of the same sample (total 3) is decreasing the number of peptides to 8,000 and inclusion of all the 18 raw files is further drastically decreases to 4,400 peptide corresponding to 1,200 proteins. Hence, the question remains the same as I have mentioned in the title of this thread.
I just compared the common and unique peptides identified from Signle and Multiple RAW file search using the same set of parameters and workflow. The comparitative venn diagram is attached below. There are 6000+ peptides fails to qualify as true peptides from multiple RAW file search. When the reason for such a loss was inspected manually in the skyline document, I get to see that there are some replicates where the peptide peak groups are picked far away from the predicted RT. Although the preicted RT range consist of a true peak group with all the transitions identified as in other replicates.
For example, the peptide "LALETALMYGAK" is getting picked only in 10 raw files out of 18 in its predicted RT range. However, when I manually check the predicted RT range in other 8 files, I can see all the transitions with similar peak shape are present but not picked or may be missed by the algorithm. Even, the corresponding decoy peptide does not have any transitions matching in the same predicted RT. The screenshot of the same are attached below for your reference.
I dont know where exactly the problem lies here. But this mismatch in peak picking is resulting with loss in the identification of many true peptide peaks.
--
Chinmaya |
|
| |
| Nick Shulman responded: |
2020-10-10 08:51 |
I would love to take a look at your Skyline document where it was crashing due to low memory. I cannot think of a reason that having a Prosit library would significantly increase memory usage, so it might be that Skyline is doing something stupid.
Whenever I need to diagnose a problem with trained model peak picking, the only thing that I know how to do is export all of the candidate peaks by doing:
File > Export > mProphet Features
(make sure that "Best scoring peaks" is not checked)
and then look at the peak that I thought Skyline should have picked, multiply its scores by the weights from the model, and then see why it did not score as well as the peak that Skyline did pick.
After you have a trained model with weights, the peak picking for each peptide in each replicate will be independent from each other. So, you can delete all of the peptides except for the one that you are investigating (although you have to keep your iRT peptides in your document), and you can remove all of the other replicates too. This might make it easier for you to examine the exported mProphet features file.
-- Nick |
| |
| Chinmaya k responded: |
2020-10-14 02:22 |
The skyline document with Human Spectral Library from Prosit which fails to imprt the raw data is uploaded to the file sharing folder under the description "Human_Prosit_Lib_Search_Skyline_Document". The import of single raw file with option "Many" under Files to import simultaneouly works if I keep it for overnight. Whereas, Skyline does not respond forever if I import with "Several" option. I have tried with command line version of Skyline yet, I dont know how best it helps in dealing with such large data analysis. I have also included a DIA raw file for your trials in the zip file shared. The system configuration is - Intel Core i9-9900KF CPU @ 3.6GHz Processor, 48 GB RAM with 300GB space in the drive.
I have exported the feature scores for peptide "LALETALM(+15.9949)YGAK" inot two files from the Skyline document after mProphet reintegration. Peptide feature scores from files where it was falsely picked were exported inot file COPD_DIA_25Da_PI_6_3_0.02Da_Final_100620_LALWTALMYGAK_False_Peaks_Picking_Specific_Files_mProphet_Features_101320.csv. Whereas, the peptide features scores from files, where it was identified consistently at the same RT were exported to COPD_DIA_25Da_PI_6_3_0.02Da_Final_100620_LALWTALMYGAK_True_Peaks_Picking_Specific_Files_mProphet_Features_101320.csv. Both the csv files and a file with weights for each feature score are attached below. The mProphet feature score files have weight multiplied values as well. As per my observation based on the weight multplied scores, the peptide "LALETALM(+15.9949)YGAK" was not picked at right RT in some of the files because of low Precursor-product shape score, Precursor mass error and Precursor isotope dot product. However, I could not find any other feature score is showing any significant deviation. If I have missed anything, please let me know.
--
Chinmaya |
|
| |
| Nick Shulman responded: |
2020-10-14 05:03 |
Thank you for sending me that file "DIA_25Da_PI_6_3_0.02Da_Human_Prosit_Lib_Search.sky.zip".
I see that you have 500,000 peptides and 6 million transitions. (half of those are decoys).
When you are extracting chromatograms, the amount of memory used is proportional to the number of transitions times the number of result files being extracted at once.
If Skyline ever needs to use more memory than the amount of RAM in the computer, then things slow down by a factor of about a million. This has to do with the way that garbage collection works, where every piece of data that the program is using needs to be periodically examined, and since it cannot all fit in memory at once, it gets into a cycle of having to read and write to disk an enormous amount of times.
The .zip file that you uploaded was missing the file "Human_RefSeq109_Prosit_Library_NCE30.blib". If you use the menu item "File > Share" to create your .sky.zip file, it will include all of the necessary files.
The setting for "How many files to import simultaneously" has no effect if you have only told Skyline to import one result file. That setting only makes a difference if you have selected multiple files in the Import Results Files file picker window.
Skyline seems to need about 20GB of RAM to do the chromatogram extraction for one file. You should choose "One at a time" for how many to files to import simultaneously, since each additional file being imported simultaneously will add about 16GB to the memory usage.
Can you send me your .sky.zip that you got your screenshot from?
If you don't want to send such a big file you could make a smaller Skyline document by deleting all of the peptides except for LALETALMYGAK and your iRT standards. Then, in order to shrink the size of the .skyd file, you need to go to Edit > Manage Results > Minimize and choose "Remove Unused Chromatograms".
-- Nick |
| |
| Chinmaya k responded: |
2020-10-14 23:58 |
Then I can remove peptide precursors with charge 5 and 6, since there are quite a few of them in the data we acquire. I guess, this should help in reducing the number of transitions and importing raw data without any memory issues to some extent.
A new skyline document with description "Human_Prosit_Lib_Search_Skyline_Document_02" is uploaded to your shared folder, which consists of complete spectral library used.
I have also uplaoded another skyline document specific to peptide "LALETALMYGAK" with iRT standards as you have mentioned. This document is available with description "LALETALMYGAK_XIC_mProphet_101520".
--
Chinmaya |
| |
| Nick Shulman responded: |
2020-10-15 03:41 |
Thank you for sending those files.
The peptide LALETALMYGAK was essentially not detected in any of your replicates.
You can use the Results Grid or the Document Grid to look at the Detection Q Value for peptides that you are interested in. If the Detection Q Value is above .05, then there is very little evidence that the peptide is present.
It might be a good idea to tell Skyline not to integrate these low confidence peaks so that you do not see them. You can do that by going to:
Refine > Reintegrate
and select "Only integrate significant q values".
-- Nick |
| |
|
|
 Peptide_Setting.PNG
Peptide_Setting.PNG Edit_iRT_Calculator.PNG
Edit_iRT_Calculator.PNG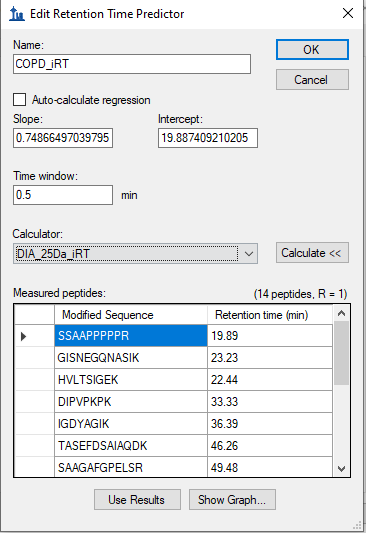 Edit_Retention_Time_Predictor.PNG
Edit_Retention_Time_Predictor.PNG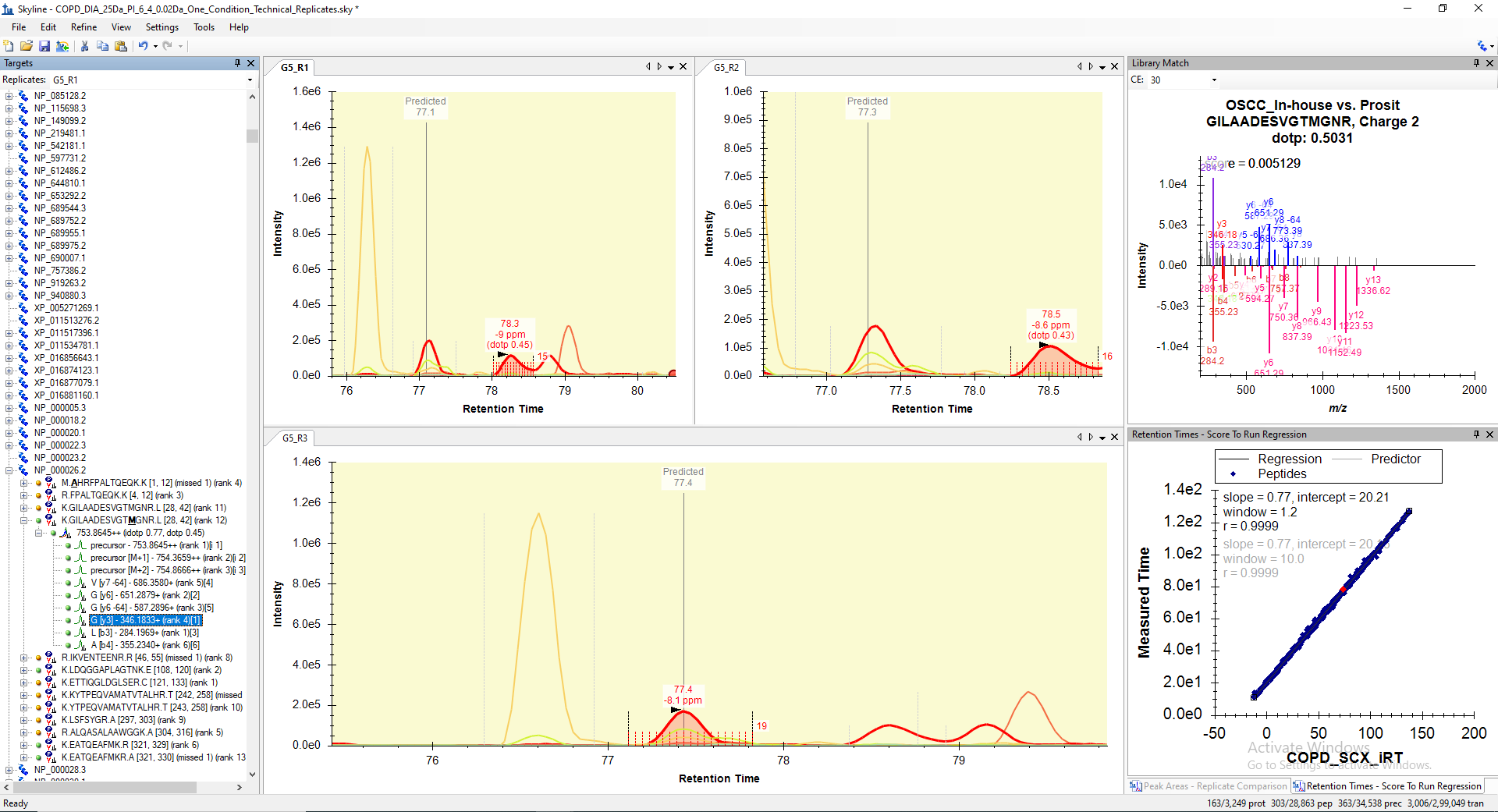 Missing_Peptide_GILAADESVGTMGNR_Inaccurate_RT_Detection.PNG
Missing_Peptide_GILAADESVGTMGNR_Inaccurate_RT_Detection.PNG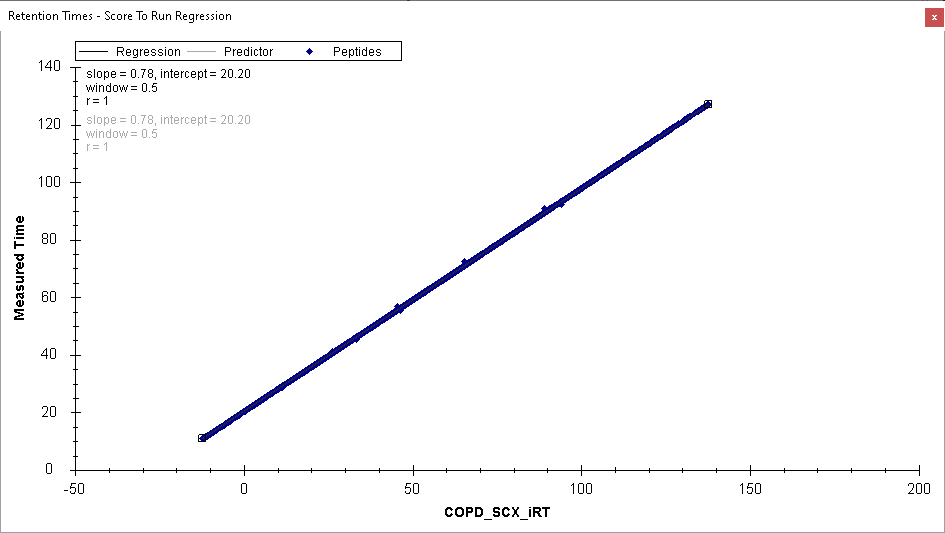 SingleFileRetentionTimesScoreToRunRegression.png
SingleFileRetentionTimesScoreToRunRegression.png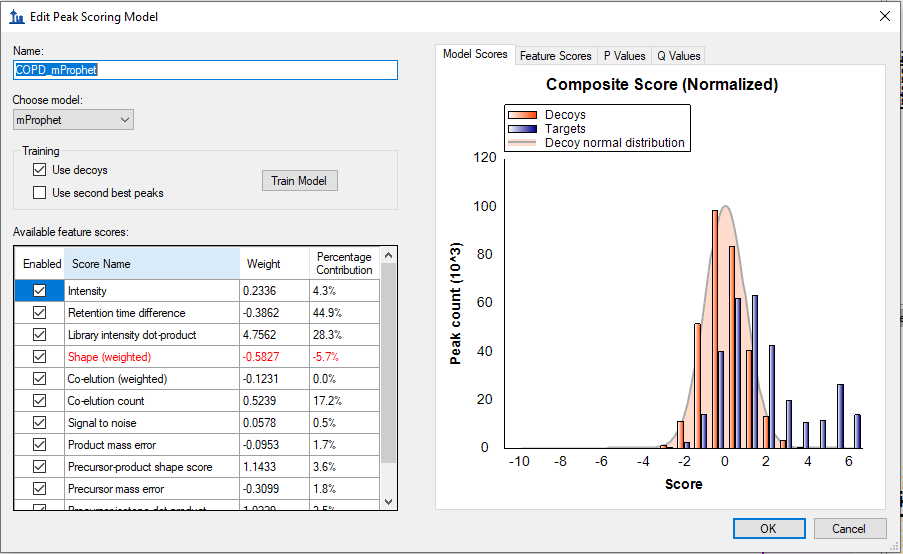 mProhet_Feature_Scores.PNG
mProhet_Feature_Scores.PNG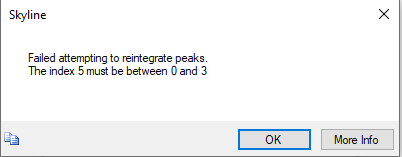 mProphet_Reintegration_Error.PNG
mProphet_Reintegration_Error.PNG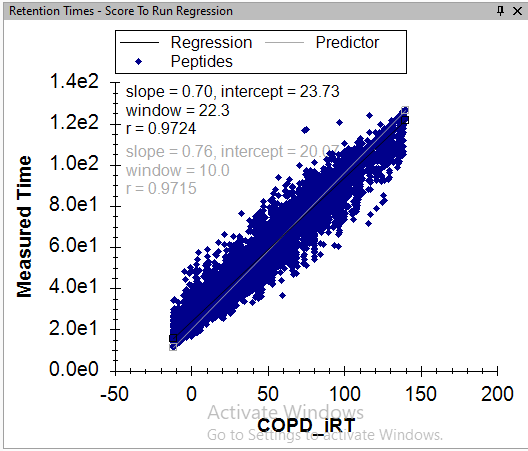 Score_to_Run_Regression.PNG
Score_to_Run_Regression.PNG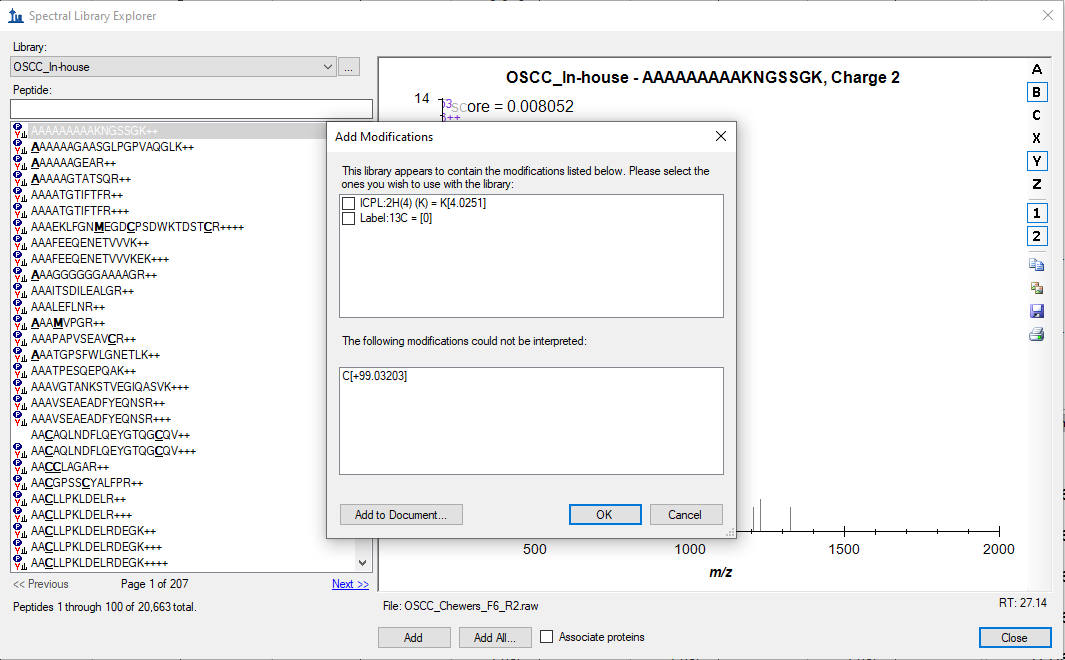 Spectral_Library_Explorer.PNG
Spectral_Library_Explorer.PNG Common_and_Unique_Peptides_from_Single_and_Multiple_RAW_file_Search.png
Common_and_Unique_Peptides_from_Single_and_Multiple_RAW_file_Search.png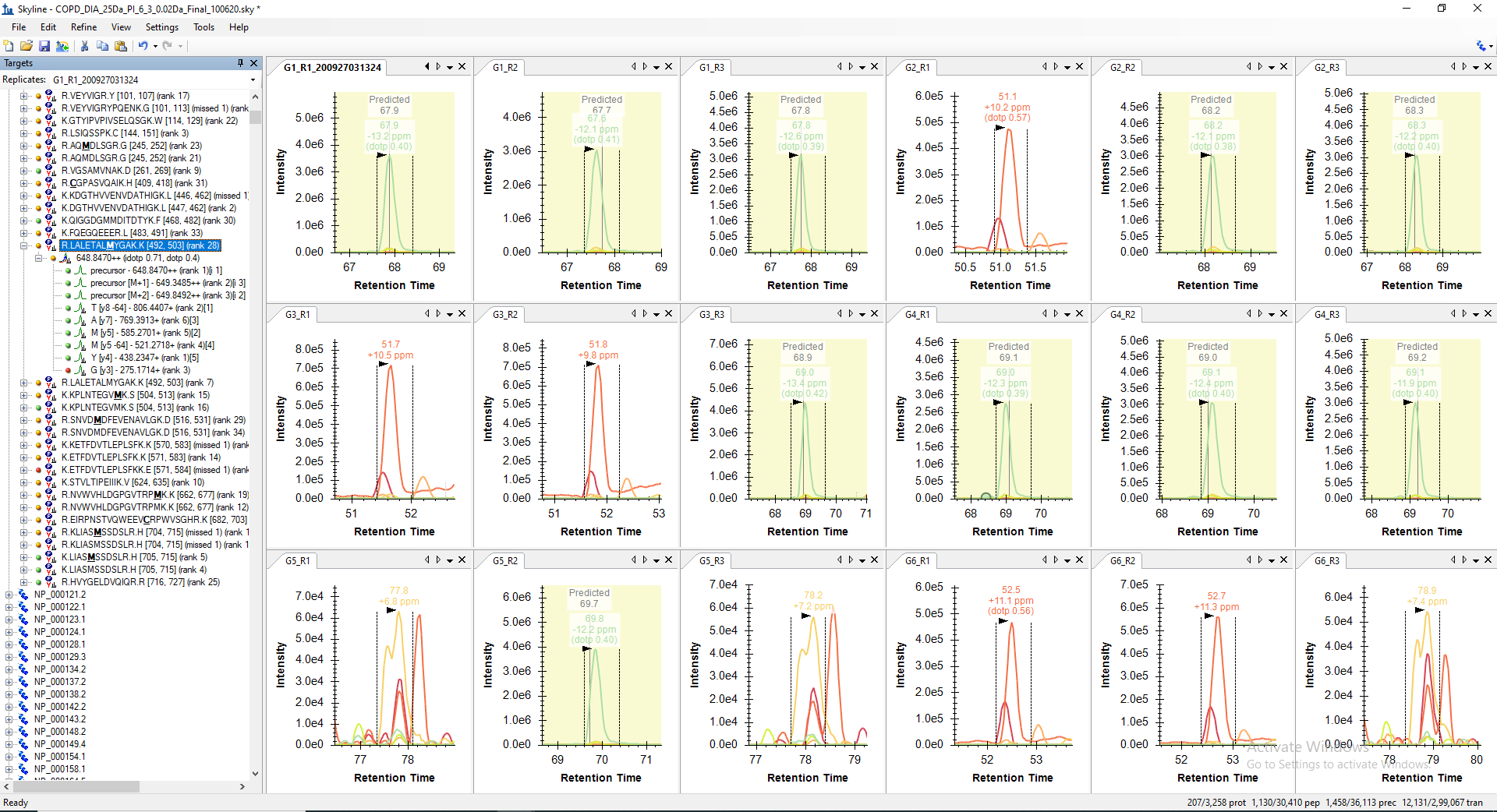 LALETALMYGAK_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files.PNG
LALETALMYGAK_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files.PNG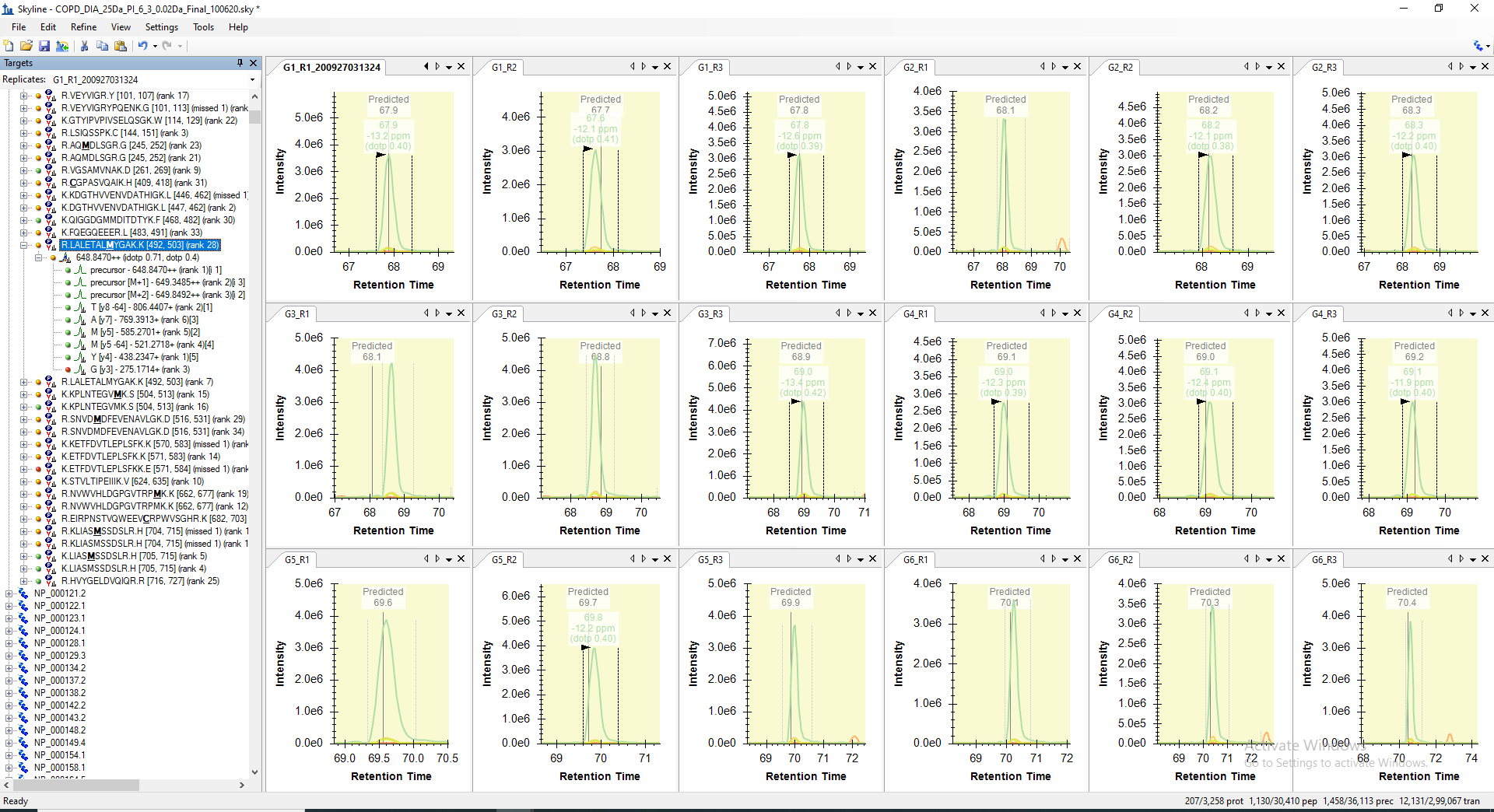 LALETALMYGAK_Manual_Visualization_Peakgroups_All_18_RAW_Files.PNG
LALETALMYGAK_Manual_Visualization_Peakgroups_All_18_RAW_Files.PNG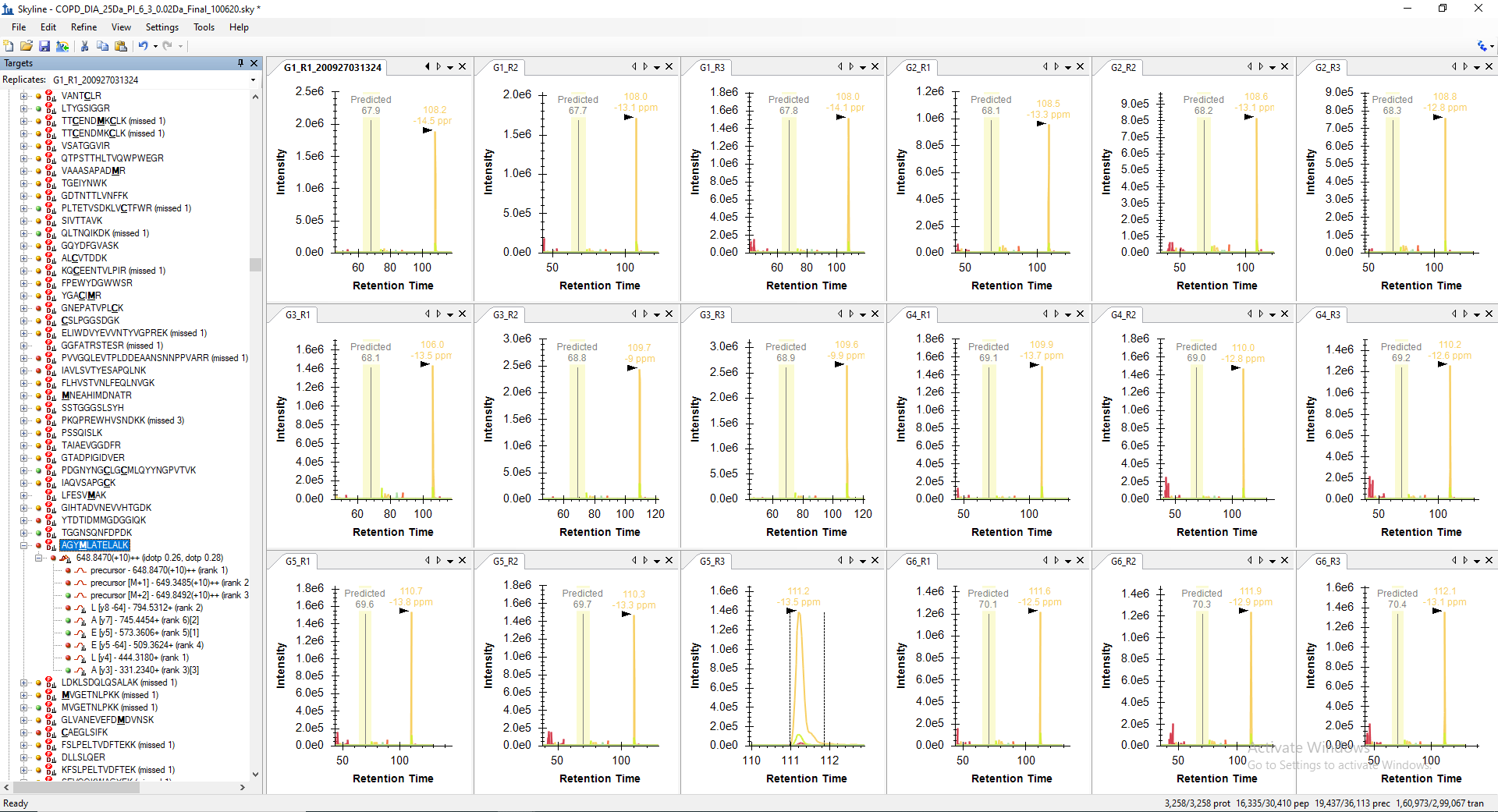 LALETALMYGAK_Decoy_Peptide_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files.PNG
LALETALMYGAK_Decoy_Peptide_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files.PNG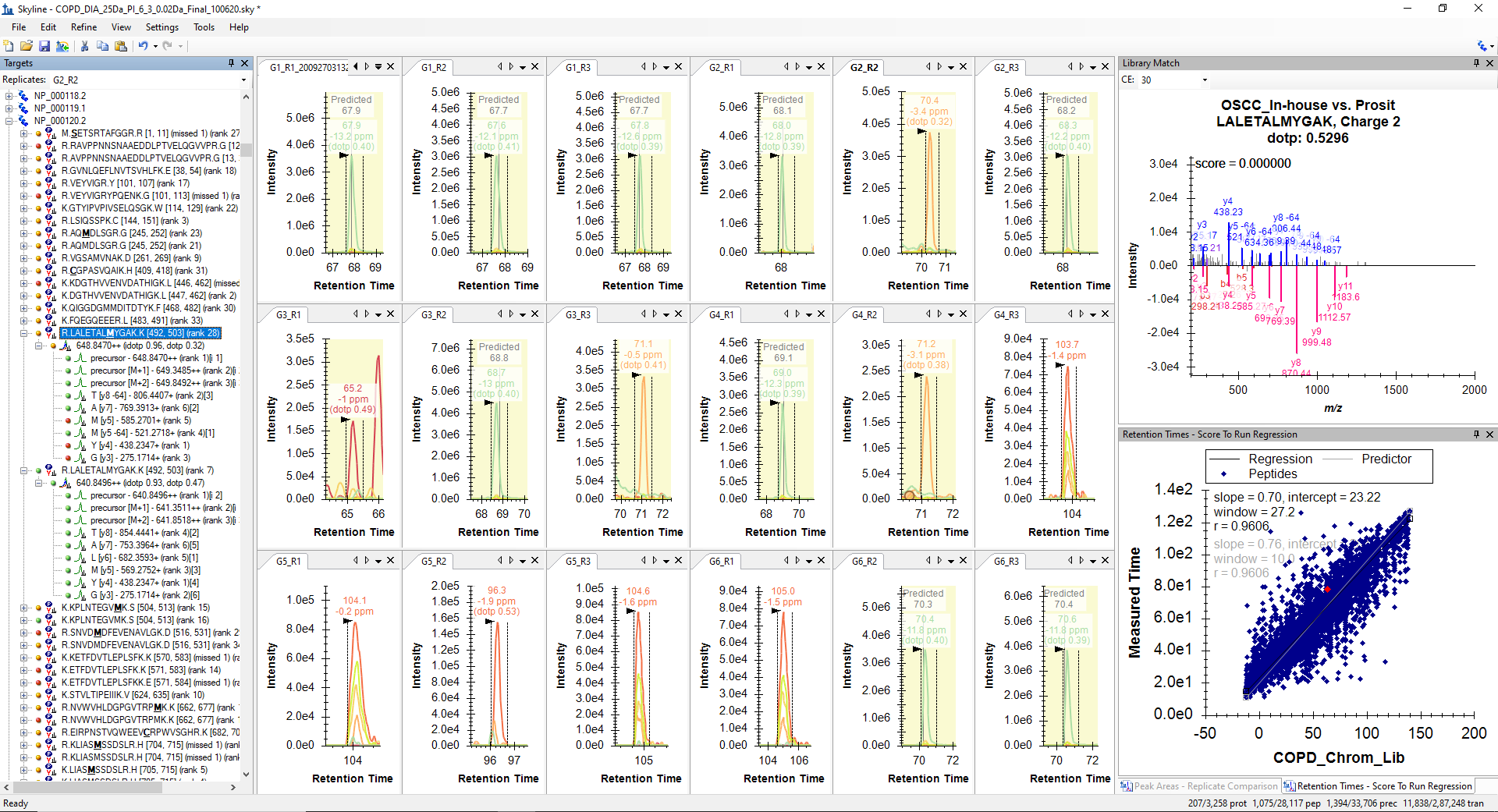 LALETALMYGAK_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files_101320.PNG
LALETALMYGAK_RT_calibrated_mProphet_Reintegrated_Peakgroups_All_18_RAW_Files_101320.PNG