Jesse,
The .sky.zip file that you attached seems to be missing the .skyd file (which contains the extracted chromatograms).
Can you try creating that .sky.zip again and send it to us?
If that .zip file is less than 50MB you can attach it to this support request. Otherwise, you can upload it here:
https://skyline.ms/files.url
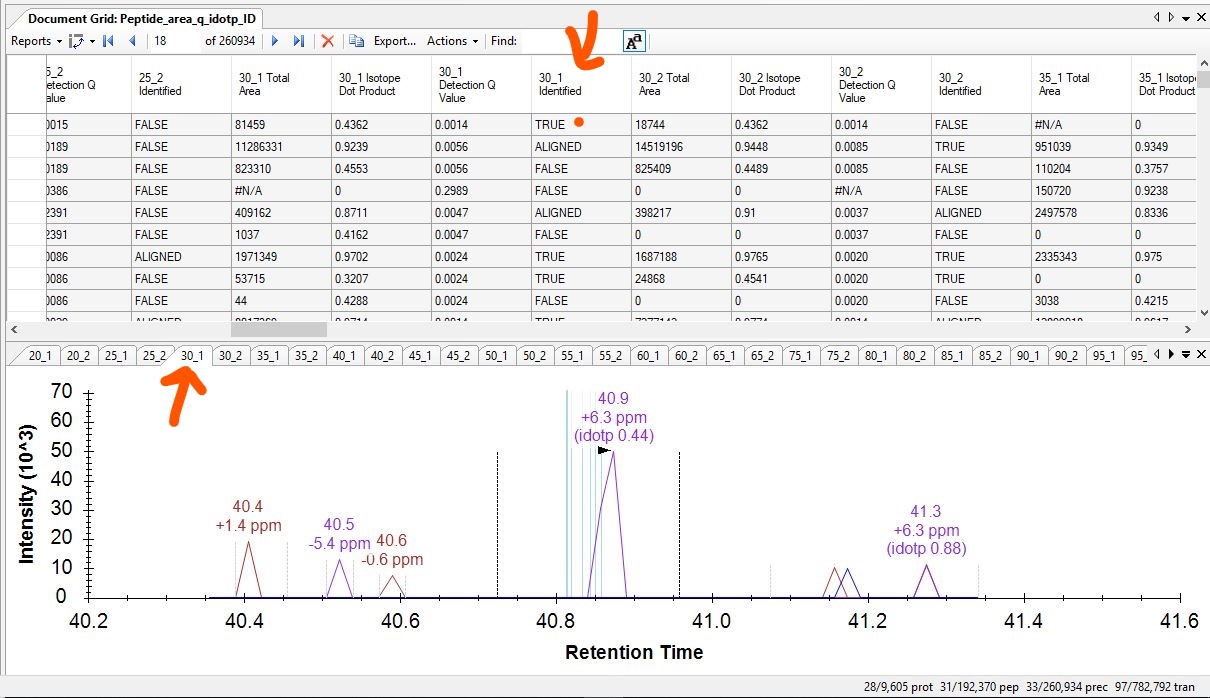
By the way, the "Identified" column in the document grid tells you whether there was an MS/MS identification between the start and end times of the chosen peak. So, we would definitely expect that if you moved to peak to some other part of the run where there were no IDs, that would change to "FALSE".
Skyline shows the location of the MS/MS IDs on the chromatogram in two ways. Identifications that happened in the file that the chromatogram came from are shown in dark blue, with the word "ID" at the top. Your screenshots do not show any of those matching ID's, but that might be because you need to right-click on the chromatogram and turn on "Peptide ID Times > Matching".
Skyline also shows MS/MS IDs that happened in other runs, but which were mapped to the times in the chromatogram's run using retention time alignment. Those aligned ID times are shown as light blue lines like in your screenshot.
The "Identified" column in the chromatogram tells you whether there was an MS/MS ID within the integration boundaries, and it will be either "TRUE", "ALIGNED" or "FALSE".
I do not believe that there is an easy way to find out whether the peptide was identified anywhere along the chromatogram, regardless of the chosen integration boundaries.
BiblioSpec spectral libraries (.blib) files are SQLite databases, so, if you are familiar with database tools, you could query this information by JOINing the RefSpectra, RetentionTimes, and SpectrumSourceFiles tables.
-- Nick
 Inkedwrong ID 1 skyline_LI.jpg
Inkedwrong ID 1 skyline_LI.jpg wrong ID 2 skyline.JPG
wrong ID 2 skyline.JPG