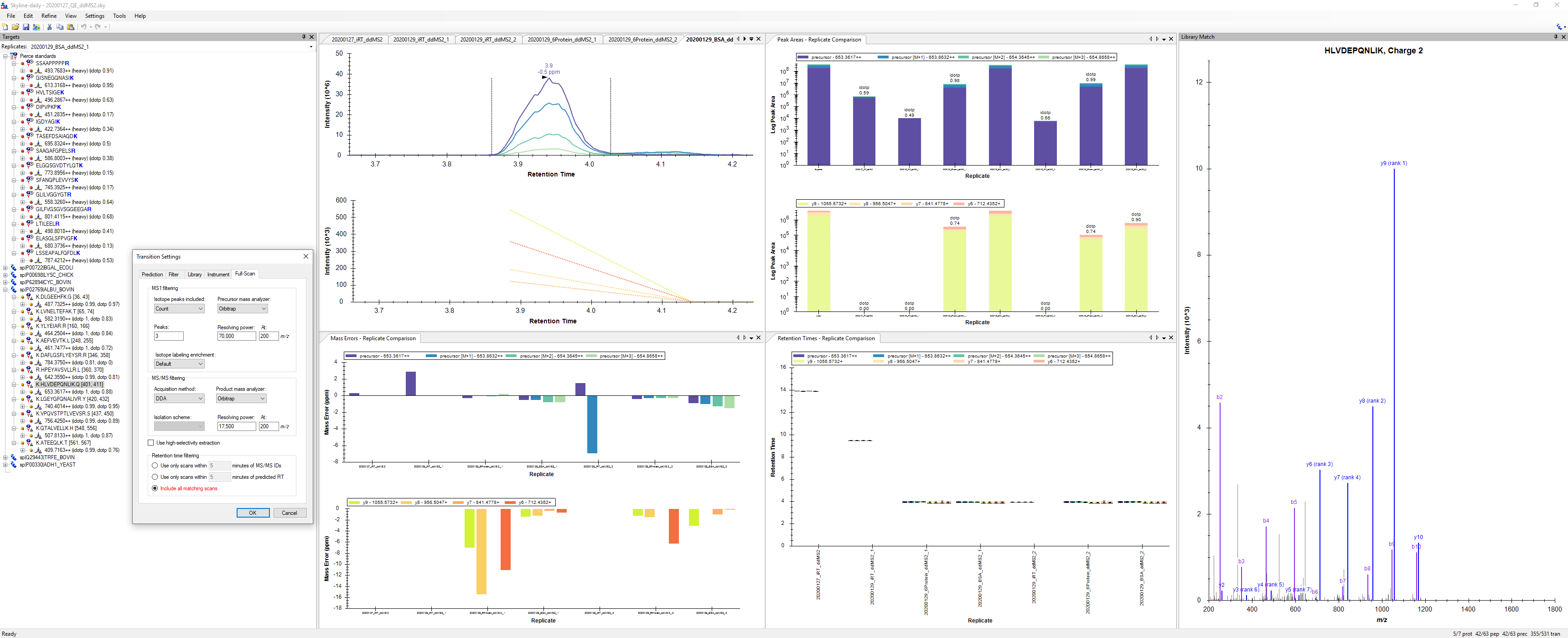
In a DDA experiment, there is no regular schedule on which particular precursors get selected by the mass spectrometer for fragmentation. Because of this, a typical DDA MS2 chromatogram will look like what you have with long straight lines connecting the times when a precursor with the same m/z as your peptide was sampled.
When you tell Skyline that your MS2 acquisition is DDA, a few things happen:
1. Skyline treats all of your MS2 transitions as non-quantitative. That means that Skyline does not include their areas under the curve in the precursor's Total Area, etc. Also, the chromatograms are displayed as dotted lines indicating that they are non-quantitative.
2. Skyline is a little more liberal about which MS2 spectra contribute to a particular precursor's chromatogram. In a DIA or PRM experiment, the precursor's monoisotopic m/z needs to be within the MS2 scan's isolation window. If the acquisition method is DDA, then scans that match any of the isotopes of the precursor (mass plus one, plus two, etc) also get included in the chromatogram.
3. Skyline ignores the MS2 chromatograms when doing peak finding.
Your data looks like what we would expect for a DDA experiment.
Is there something that you would expect to be or behave differently?
-- Nick |
Hi Nick,
Thanks for the explanation, this is very helpful! We were just comparing DIA with DDA as a POC kind of work, so it is good to know this is the way the data is supposed to look.
Thanks again,
Ana |
 ddMS2 data_20200130AI.png
ddMS2 data_20200130AI.png