| Brendan MacLean responded: |
2019-11-27 22:04 |
Hi Arman,
Please post a screenshot of your Transition Settings - Full-Scan tab. It could be that you have chosen too narrow tolerance for your chromatogram extraction.
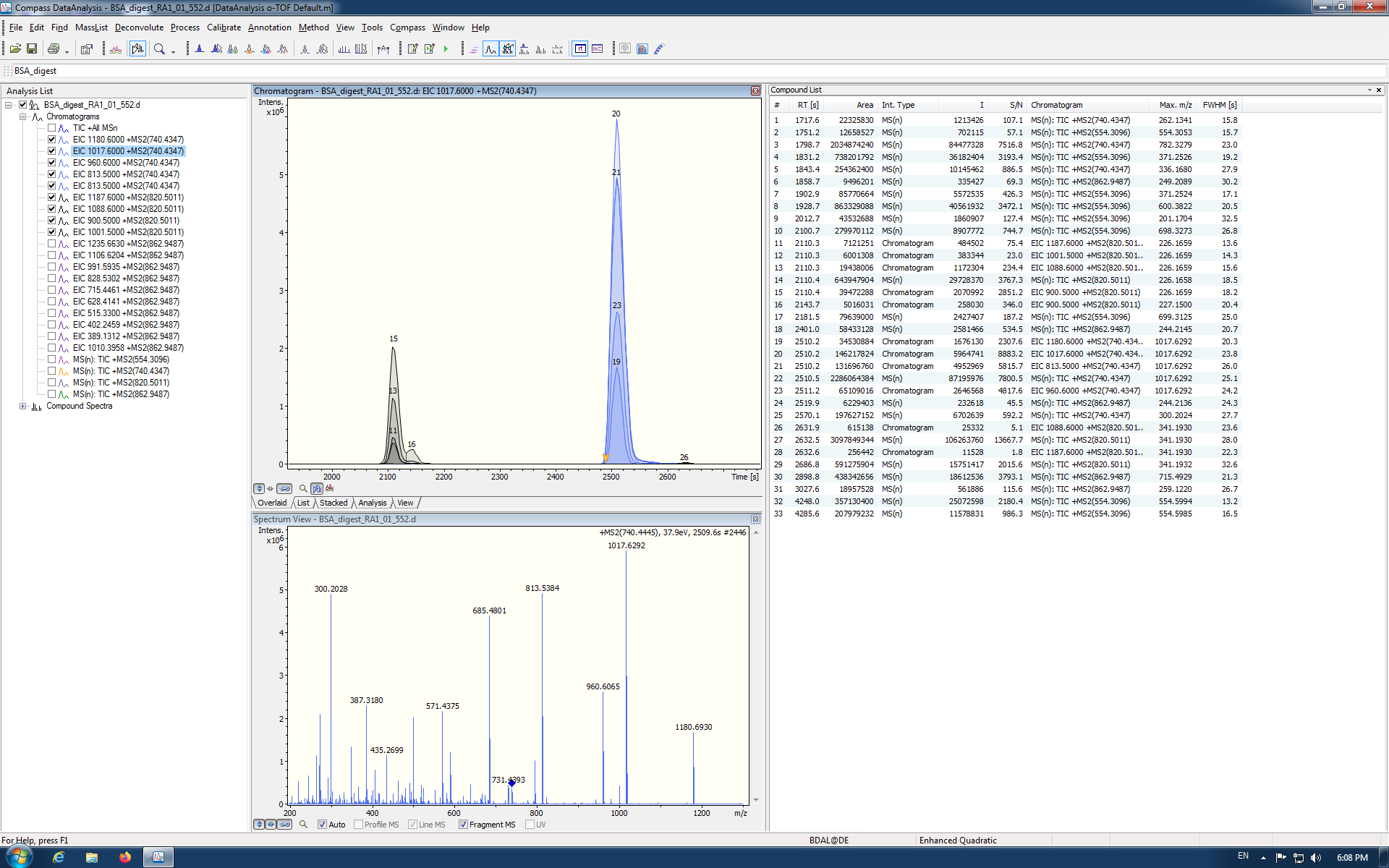
Also, unless I am missing something the masses are not the same:
- 820.4725 in Skyline
- 1088.5946
- 900.5148
- 325.2234
- 820.5011 in DataAnalysis
- 1187.600
- 1088.600 *
- 900.500 *
- 1001.500
Remember that 0.01 is 10 ppm at 1000. So, if you have a 10 ppm mass tolerance for Centroided extraction, you may be extracting from disjoint ranges between 900.5148 and 900.500, which differ by 0.0148.
If you are using a "MRM" (a.k.a. PRM) method, I couldn't tell you whether the difference of 0.0286 in the precursor m/z values makes any difference. Skyline still may be matching the same MS/MS spectra for extraction. I guess my sense is that the 820.5011 value in DataAnalysis is truly what the instrument collected, but this is off by 30 ppm or so from what Skyline calculates for the true m/z of the peptide. This would make a bigger difference, if you were attempting to extract precursor ions from high-resolution MS1 spectra, but Skyline does seem to be matching the precursor to MS/MS spectra you have collected. So, I suspect the key to this issue lies in the product m/z values and the MS/MS filtering settings in Skyline.
The extraction time does appear to be right. 35 minutes corresponds to the peak at 2100 seconds in your DataAnalysis screenshot.
Please send that screenshot of your Full-Scan tab. Thanks for the helpful screenshots.
--Brendan
|
| |
| akulyyasov responded: |
2019-11-28 02:10 |
Dear Brendan,
Thank you very much for the explanation of this problem.
I attached screenshots of Full-Scan tab and also Edit Isolation Scheme tab where I imported isolation windows from the raw file.
With best regards,
Arman
|
|
| |
| Brendan MacLean responded: |
2019-11-28 06:08 |
So, first, you don't want to use a DIA isolation scheme, because you are doing "MRM" (a.k.a PRM), and that isolation scheme you got from using import isolation scheme is not meaningful for PRM. So, switch your acquisition method to "Targeted", which I have been strongly considering changing to PRM. Would you have chosen that then? Also since you are not targeting any precursor ions in your transition list that I can see, you should turn off MS1 filtering, unless you want to add some precursor ions to target in the MS1, in which case, you should add "p" to the ion types in your Transition Settings - Filter tab.
Also, my understanding is that it is really best to use mass analyzer = "Centroided" with Bruker QTOF data, but I would start off with 20 ppm until you prove to yourself that the data can support a narrower extraction range. Your current MS/MS setting of mass analyzer = QTOF with 30,000 resolving power is quite wide (around 50 ppm). So, it is not likely that you are missing signal that is present, but it is still possible that your chromatograms are getting extracted from the wrong spectra, due to your DIA settings.
I have attached settings I think might be most appropriate from what I have seen and what you have told me. Give these a try and let me know how they work. They may not solve your problem entirely, but they should change things and we can go from there.
--Brendan
|
|
| |
| akulyyasov responded: |
2019-11-30 01:55 |
Dear Brendan,
Thank you very much for your help and the attached settings. The extracted ion chromatograms slightly improved but still are not so perfect like in the DataAnalysis program. Please find attached the png file of these results with new settings.
With best regards,
Arman
|
|
| |
| Brendan MacLean responded: |
2019-11-30 06:39 |
Once you have peaks this good, you can click on the peaks in the chromatograms you see and look a the underlying MS/MS spectra from which Skyline is extracting. This is best explained in our DIA tutorial in the "Understanding Extracted Chromatograms" section on page 31:
https://skyline.ms/_webdav/home/software/Skyline/@files/tutorials/DIA-2_6.pdf#page=31
You can use this feature of Skyline to understand why Skyline may not be ending up with XICs like you expect, or to convince yourself that the XICs do or do not accurately represent the underlying spectra. If they do, I can't really explain why DataAnalysis ends up with something different.
In your plot, it looks like you might want to reimport your data file, because you have targeted as many as 8 to 12 transitions, but only have XICs for the top 3, which is all you were targeting in your original screenshots. When you change the transitions, you will need to reimport to get new XICs.
|
| |
| akulyyasov responded: |
2019-12-02 22:47 |
Dear Brendan,
I clicked on the peaks in the chromatogram and I see MS/MS spectra. I also checked that the retention time information is in seconds (Bruker DataAnalysis 4.3 (x64)). Should I always change RT from min to s before passing to Skyline? In Bruker OTOF control software they use another terminology for targeted methods. As far as I understand middle-band can be used for MRM (or PRM) and broad-band for DIA? When I run the sample I modified Bruker's template method (Targeted protein quantification middle-band CID-MRM.m file) for 4 precursors 554.3096, 740.4347, 820.5011 and 862.9487 with width 3.0 and in Edit isolation scheme tab, I imported these values from the raw file.
With best regards,
Arman
|
| |
| akulyyasov responded: |
2019-12-09 00:49 |
Dear Brendan,
I repeated again passing data from analysis of BSA sample to Skyline with parameters in Full-Scan Tab as you recommended before. I see perfectly coeluted extracted ion chromatograms for the precursor ion 740 which is similar to the DataAnalysis EIC. But why EIC for the ion 820 is absent in Skyline?
With best regards,
Arman
|
|
| |
| Brendan MacLean responded: |
2019-12-09 06:46 |
Hi Arman,
I don't understand. I don't see in your screenshot where you are targeting a precursor with m/z of 820. Can you clarify what you are expecting to see in your Skyline screenshot and why it captures something unexpected? I am missing the point.
Thanks for the update. At least it shows a very nice peak for 740, as you say, but you will need a target for an 820 precursor to see a peak for 820. The screenshot shows only 740, 862, and 554, but only the 740 peak.
--Brendan
|
| |
| akulyyasov responded: |
2019-12-10 00:30 |
Dear Brendan,
Now I imported results where BSA analysis data were analyzed and saved in the format with retention time expressed in minutes.
Before I always change RT to seconds in option in the menu of DataAnalysis program (Bruker DataAnalysis 4.3(x64)) as was recommended by Application Tutorial of Skyline.
It is strange, but in this case, I see a perfectly coeluting peak at 820 similar to the peak obtained in the DataAnalysis program.
Does it mean that it is not necessary to change RT from minutes to seconds?
With best regards,
Arman
|
|
| |
| Brendan MacLean responded: |
2019-12-10 07:47 |
Hi Arman,
You have lost me. I don't know of the "Application Tutorial of Skyline" you refer to, and the last screenshot you posted was showed chromatograms for 740 and not 820. The peak looked fine. Though it was not at the time one would expect for 820. But the precursor was 740.
Anyway, if you are now satisfied, then I suggest you continue with what you have worked out.
Good luck with your research. Thanks for using Skyline with your Bruker data.
--Brendan
|
| |
 DataAnalysis_BSA.png
DataAnalysis_BSA.png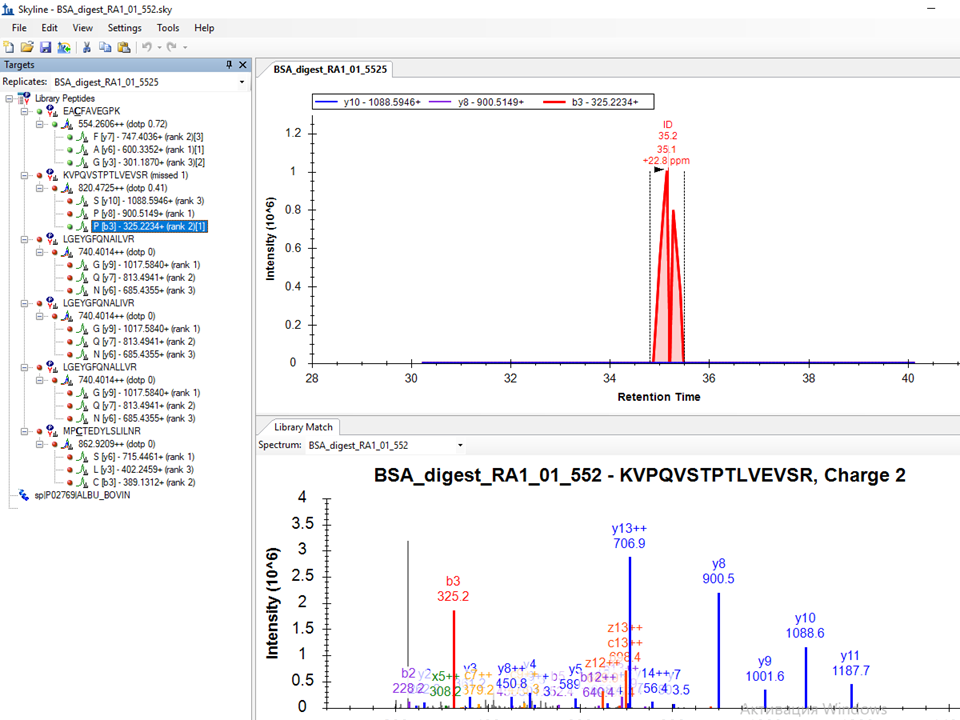 Skyline_BSA.png
Skyline_BSA.png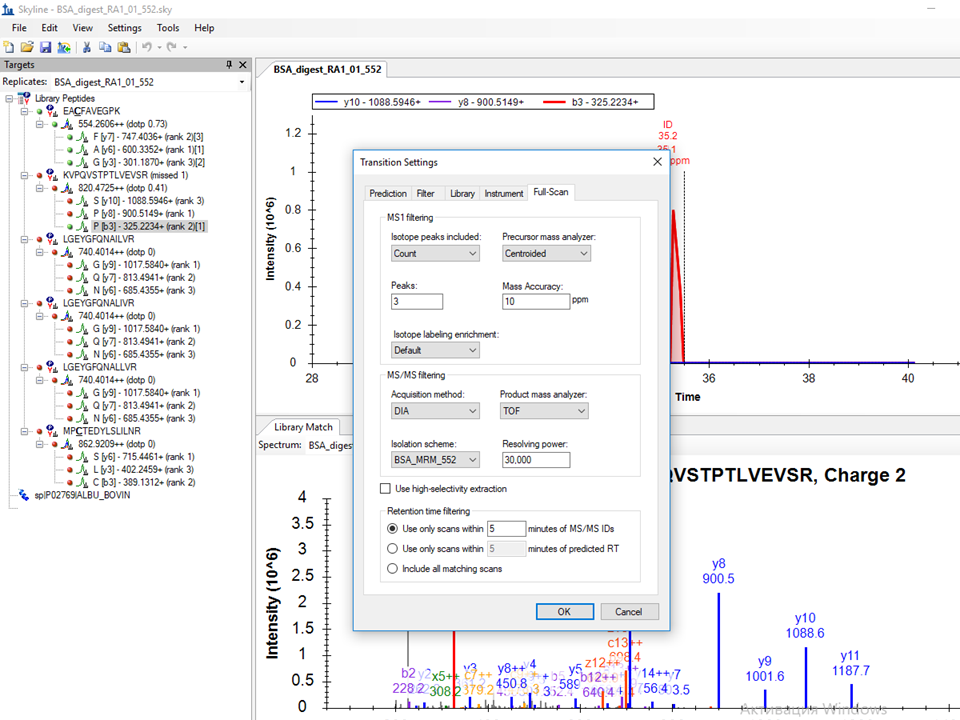 Skyline_BSA_Full Scan Tab.png
Skyline_BSA_Full Scan Tab.png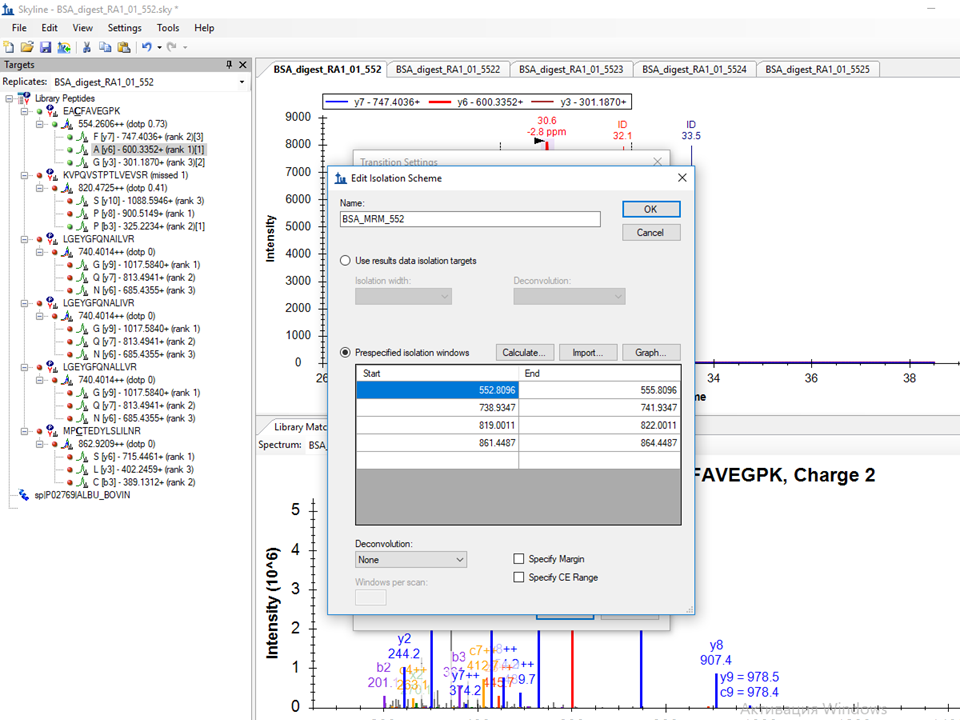 Skyline_BSA_Edit Isolation Scheme.png
Skyline_BSA_Edit Isolation Scheme.png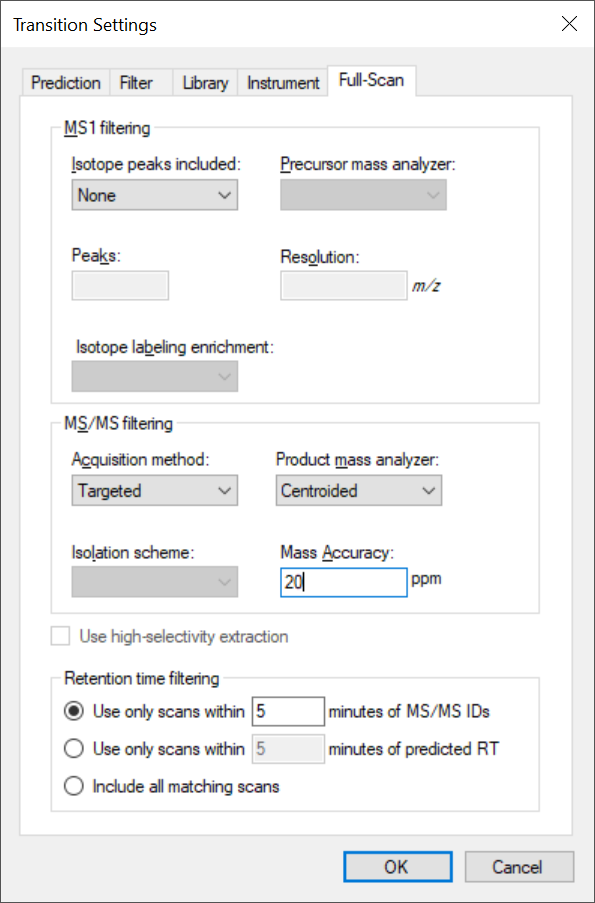 Bruker PRM Full-Scan Settings.png
Bruker PRM Full-Scan Settings.png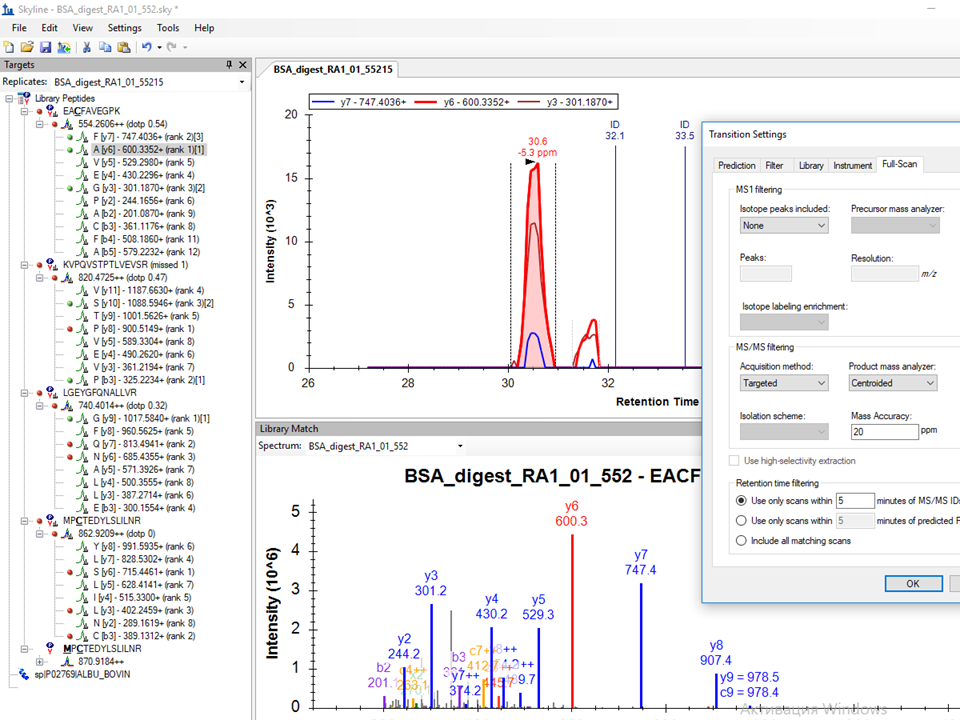 Skyline_BSA_Full Scan Tab_targeted_centroid.png
Skyline_BSA_Full Scan Tab_targeted_centroid.png BSA_552_absent peak at 820.png
BSA_552_absent peak at 820.png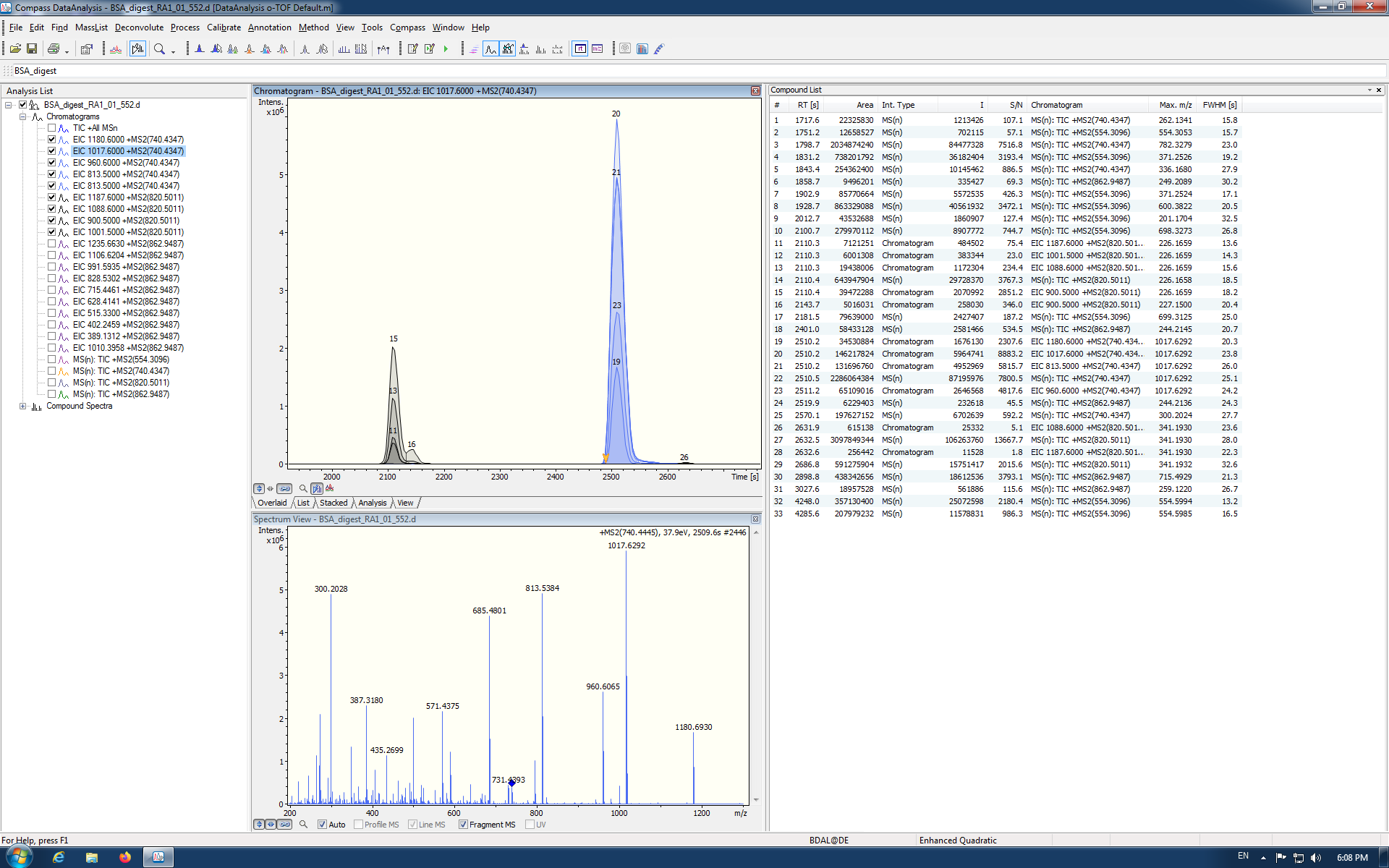 DataAnalysis_BSA.png
DataAnalysis_BSA.png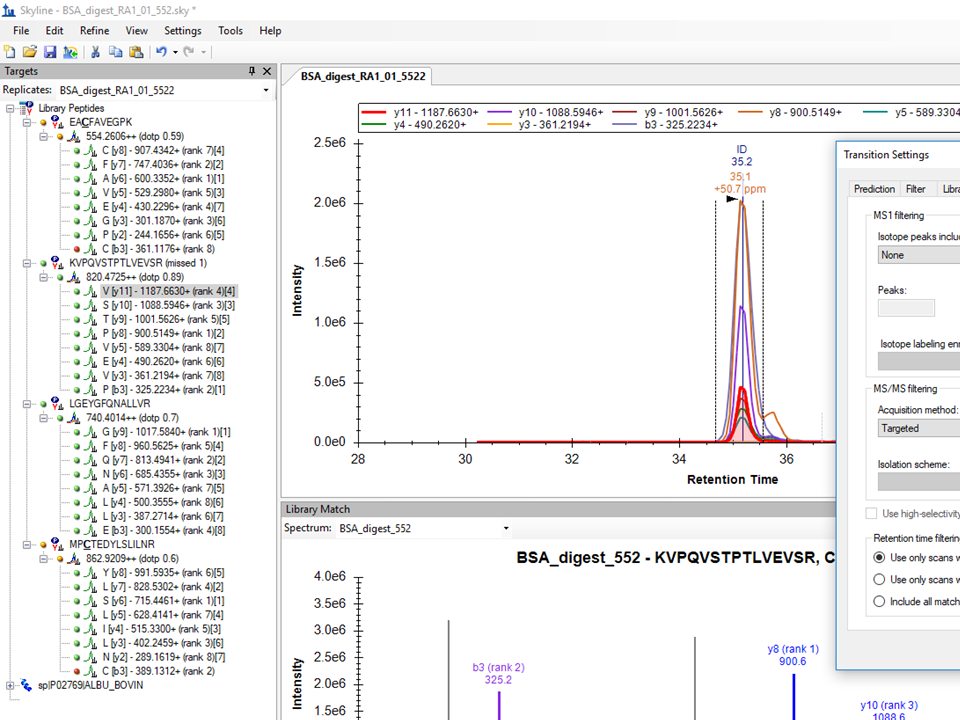 BSA_552_peak at 820.png
BSA_552_peak at 820.png